


IF-027 - PSEUDOGRANULOMAS EN INMUNODEFICIENCIA COMÚN VARIABLE
Servicio de Medicina Interna. Hospital Universitario Central de Asturias. Oviedo (Asturias).
Objetivos: La inmunodeficiencia común variable hace referencia a un grupo de síndromes caracterizados por disminución de inmunoglobulinas séricas, escasa producción de anticuerpos e infecciones recurrentes. Los pacientes con esta patología están predispuestos a desarrollar enfermedades autoinmunes hasta en un 20-25% de los casos. Existen casos descritos de desarrollo de granulomas no caseificantes desarrollando un cuadro similar a la sarcoidosis y que se denomina “sarcoidosis-like”.
Métodos: Descripción de dos casos diagnosticados de inmunodeficiencia común variable en los que se han objetivado granulomas no caseificantes, con un diagnóstico inicial de sarcoidosis, en seguimiento en la Unidad de Enfermedades Sistémicas y Autoinmunes del Hospital Universitario Central de Asturias.
Resultados: 1. Paciente diagnosticada en un principio de sarcoidosis en relación con la presencia de granulomas no caseificantes en adenopatías mediastínicas, junto con hepatoesplenomegalia. En su evolución múltiples infecciones respiratorias: neumonías adquiridas en la comunidad, abscesos cutáneos y diarreas, por lo que se diagnóstica de inmunodeficiencia común variable según los criterios de la ESID a los 42 años (niveles de inmunoglobulinas al diagnóstico por debajo de dos desviaciones estándar a los que correspondería por edad) (IgG 3,03 g/l (6,5-17), IgA < 0,07 g/l (0,85-4,68), IgM 0,124 g/l (0,45-2,76). Reacción adversa grave al tratamiento con inmunoglobulinas endovenosas, posiblemente en relación con presencia de anticuerpos Ig contra la inmunoglobulina A, que indica una forma severa de la enfermedad. Múltiples ingresos por infecciones respiratorias bajas llegando a precisar en alguna ocasión ingreso en Cuidados Intensivos. Ante las infecciones recurrentes, diarreas incoercibles y un % de linfocitos de memoria inferior al 1% se ha iniciado tratamiento con inmunoglobulinas endovenosas, con la mejoría de la sintomatología referida a la inmunodeficiencia humoral, y sin progresión de las adenopatías del mediastino. 2. Paciente con diagnóstico a los 30 años de inmunodeficiencia común variable siguiendo los criterios de laESID en relación con infecciones respiratorias de repetición y bronquiectasias crónicas bilaterales por Pseudomonas aeruginosa multirresistente. Presencia de placas sobreelevadas eritematosas muy induradas y con tendencia a la descamación posterior en la muñeca izquierda y en la rodilla ipsilateral, que se presentan a los dos años del diagnóstico. Biopsia compatible con lesiones granulomatosas que afectan la dermis profunda con discretos focos de necrosis central con tinción de Zielh negativa. Se ha planteado tratamiento con hidroxicloroquina, que no se ha podido iniciar, al presentar la paciente un maculopatía de origen indeterminado.
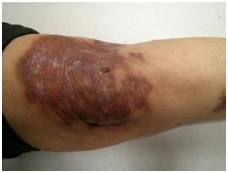
Discusión: Los granulomas tipo sarcoidosis pueden presentarse entre un 5-10% de los pacientes con inmunodeficiencia común variable. Los granulomas no caseificantes aparecen a nivel de parénquimas pulmón, hígado, riñón, ganglios linfáticos y en el ojo (uveítis granulomatosa). La disminución de las células B que no realizan adecuadamente el switch (cambio de isotipo) parece ser el mecanismo patogénico responsable.
Conclusiones: La aparición de granulomas en pacientes con inmunodeficiencia es una entidad infrecuente. Ante la aparición de granulomas sin una evolución clínica habitual de sarcoidosis, hay que excluir una inmunodeficiencia común variable.


