


IF-084 - DESCRIPCIÓN DE 6 CASOS DE SÍNDROME HEMOFAGOCÍTICO DIAGNOSTICADOS EN UN SERVICIO DE MEDICINA INTERNA
Medicina Interna. Hospital San Cecilio. Granada.
Objetivos: Describir cohorte de 6 pacientes diagnosticados de síndrome hemofagocítico en Servicio de MI.
Material y métodos: Estudio observacional descriptivo retrospectivo. Incluye pacientes diagnosticados de SH en los últimos 3 años.
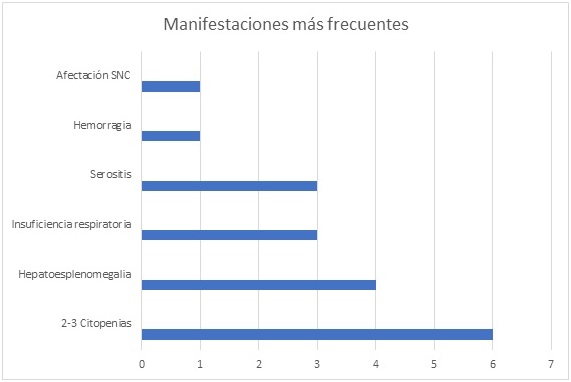
Resultados: La edad promedio fue 34 ± 15 años, siendo 83% mujeres. En la mitad de los casos, la enfermedad subyacente fue LES, siendo en un caso la forma de debut. En dos casos no se pudo determinar desencadenante, estando el caso restante relacionado con parvovirus B19. El tiempo de retraso diagnóstico fue 4 días. En el diagnóstico, todos presentaban citopenias (2-3 series), siendo frecuente hepatoesplenomegalia (66%), insuficiencia respiratoria y serositis (50%). La hemorragia y afectación SNC solo se observó en 1 caso (mayor gravedad) y únicamente detectaron adenopatías en 2 pacientes. Analíticamente, todos presentaron hiperferritinemia (1.479 ± 991 ng/ml) e hipertrigliceridemia (365 ± 146 mg/dl) con niveles de PCR al diagnóstico normales en 66% pacientes (80 ± 108 mg/dl). Un solo paciente mostró elevación de NK, sin poder cuantificar su actividad. Todos recibieron corticoides sistémicos, asociados a inmunoglobulinas endovenosas en 4 casos. La evolución fue favorable en todos los casos, sin ningún fallecimiento.
Discusión: El síndrome hemofagocítico es una entidad clínica grave y pleomórfica en cuanto a sus formas de presentación, siendo las etiologías involucradas variadas, haciendo de ella una patología de interés para el internista. Puede ocurrir por un defecto genético predisponente o desencadenante inmunológico (infecciones, neoplasias o enfermedades autoinmunes). Los hallazgos comunes incluyen fiebre, hepatoesplenomegalia, erupción cutánea, linfadenopatías, síntomas neurológicos, citopenias, hiperferritinemia y anomalías de la función hepática. El diagnóstico se realiza mediante la identificación de una mutación genética o por cumplimiento de 5/8 criterios diagnósticos (tabla). La hemofagocitosis no es necesaria ni suficiente para el diagnóstico.

Conclusiones: El diagnóstico precoz precisa de un alto nivel de sospecha. El LES fue la causa más frecuentemente en nuestra serie, suponiendo un reto diagnóstico por la superposición de sus manifestaciones. La existencia de organomegalias, hiperferritinemia y fiebre persistente deben advertir al clínico de la posibilidad de un SH secundario.
Bibliografía
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503.
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613.


