


IF-034 - SÍNDROME DE SUSAC: SERIE DE 4 PACIENTES. LA ENFERMEDAD DE GOYA
Medicina Interna. Hospital Universitario La Paz. Madrid.
Objetivos: Describir las características clínicas y tratamiento recibido de 4 pacientes diagnosticados de síndrome de Susac (sdS) en nuestro hospital.
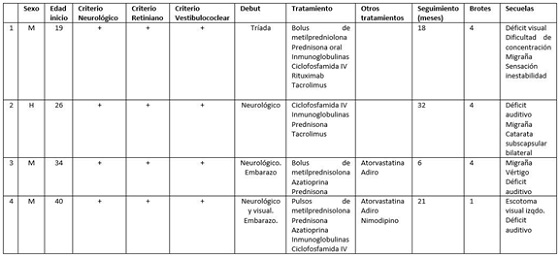
Material y métodos: Describir las características clínicas de 4 pacientes afectos: clasificación según los criterios de Kelffner 2016, edad, sexo, clínica en el debut, líneas de tratamiento, otros tratamientos complementarios, meses de seguimiento, nºde brotes tras inicio de tratamiento y síntomas residuales.
Resultados: Los 4 pacientes debutaron antes de los 40 años habiendo un mayor número de mujeres (3:1). Todos los pacientes (fig.) habían sido diagnosticados de sdS definitivo según los criterios neurológicos, retinianos y vestibulococleares (Kelffner et al, 2016). Los 4 pacientes presentaron sintomatología neurológica en el debut. Además, en 2 de las 3 mujeres el embarazo o puerperio se relacionó temporalmente con el inicio de los síntomas. Tres pacientes han recibido tratamiento inmunosupresor agresivo con ciclofosfamida o rituximab. Además, en 2 pacientes se añadieron empíricamente tratamiento con adiro, atorvastatina y nimodipino. A pesar del tratamiento inmunosupresor agresivo, los brotes de enfermedad se han presentado en un seguimiento breve en 3 pacientes. Secuelas, de diversa gravedad, se observaron en los 4 pacientes.
Discusión: El síndrome de Susac es una enfermedad rara, de posible etiología autoinmune, en la que se afectan la microcirculación del cerebro, retina y oído interno, dando lugar a una tríada sintomática característica: encefalopatía, alteraciones visuales y déficit auditivos. Recientemente se han publicado unos criterios diagnósticos y unas recomendaciones terapéuticas (Rennerbohm et al, 2018). No obstante, las series de pacientes descritas son pequeñas y no hay ensayos clínicos aleatorizados acerca del tratamiento. Todos nuestros pacientes presentaron un sdS definitivo según criterios clínicos. Las características demográficas son similares a las descritas en la literatura con una edad de debut temprana y un predominio de mujeres. Es notable la relación temporal del debut con la gestación en 2/3 mujeres. Todos los pacientes recibieron terapia inmunosupresora, 3/4 con ciclofosfamida o rituximab. A pesar de ello, los 3/4 que recibieron el tratamiento más agresivo presentaron brotes de enfermedad al menos 1 vez al año. Dado que fisiopatológicamente se trata de una vasculopatía de etiología autoinmunitaria no demostrada, se decidió añadir al tratamiento en 2/4 pacientes adiro y atorvastatina como protector endotelial. La paciente nº4 presentó la clínica neurológica más grave, por lo que se añadió, además, nimodipino. Asimismo, la comorbilidad en todos los pacientes fue importante en forma de déficit auditivo clínico o subclínico, migraña y déficit visuales con escotomas.
Conclusiones: El sdS se trata de una vasculopatía de presumible etiología autoinmune que afecta principalmente a mujeres jóvenes. A pesar del tratamiento agresivo los síntomas tienden a recurrir, asociando una comorbilidad importante. Todavía están por explorar nuevas terapias complementarias a la inmunosupresión convencional.


