





Revisión de la evidencia científica sobre el tratamiento oral de pacientes adultos con enfermedad de Gaucher tipo 1 (EG1), con formato de guía clínica, según la normativa Agree II. Se describen las principales diferencias entre los 2 tratamientos orales disponibles actualmente para el tratamiento de esta entidad (miglustat y eliglustat).
En esta revisión se recuerda que los criterios para iniciar el tratamiento oral en los pacientes con EG1 deben valorarse de forma individualizada. Si bien miglustat y eliglustat son inhibidores de la enzima glucosilceramida sintetasa, los 2 presentan diferentes mecanismos de acción y propiedades farmacológicas y nunca se deben considerar como equivalentes. Miglustat está indicado en pacientes con EG1 no grave que no pueden recibir otro tratamiento de primera línea, mientras que eliglustat está indicado en pacientes con EG1 con cualquier gravedad, en primera línea y sin necesidad de estabilización previa con tratamiento de reemplazo enzimático. Es importante enfatizar que para iniciar tratamiento con eliglustat debemos conocer el fenotipo metabólico CYP2D6 y que su asociación con fármacos metabolizados a través de los citocromos CYP2D6 y CYP3A4 –o bien que utilicen la glucoproteína P– se debe evaluar individualmente. Durante el embarazo se debe evitar el uso de eliglustat, pudiéndose emplear únicamente el tratamiento de reemplazo enzimático. A diferencia de miglustat, cuyos efectos adversos han limitado su utilización, eliglustat no solo ha demostrado una eficacia similar a la del tratamiento de reemplazo enzimático, sino que ha demostrado mejoría en la calidad de vida de los pacientes EG1.
This work is a review of the scientific evidence on the oral treatment of adult patients with Gaucher disease type 1 (GD1) with a clinical guideline format according to the Agree II regulations. It describes the main differences between the 2 oral treatments currently available for treating this disease (miglustat and eliglustat).
This review reminds us that the criteria for starting oral treatment in patients with GD1 must be assessed individually. Although miglustat and eliglustat are both glucosylceramide synthase enzyme inhibitors, they have different mechanisms of action and pharmacological properties and should never be considered equivalent. Miglustat is indicated in patients with non-severe GD1 who cannot receive other first-line treatments, while eliglustat is indicated as first-line treatment for patients with GD1 of any severity without the need for prior stabilization with enzyme replacement therapy. It is important to emphasize that in order to start treatment with eliglustat, we must know the CYP2D6 metabolic phenotype and its association with drugs metabolized through the CYP2D6 and CYP3A4 cytochromes –or alternatively those that use P-Glycoprotein– must be evaluated on an individual basis. During pregnancy, the use of eliglustat should be avoided; only enzyme replacement therapy can be used. Unlike miglustat, whose adverse effects have limited its use, eliglustat has not only demonstrated similar efficacy to enzyme replacement therapy but has also been shown to improve the quality of life of patients with GD1.
La enfermedad de Gaucher (EG) es una patología de depósito lisosomal de herencia autosómica recesiva, producida por mutaciones en el gen GBA que conducen a un defecto de síntesis o alteración funcional de la proteína glucocerebrosidasa (GCasa). Este déficit enzimático produce el acúmulo de su sustrato, la glucosilceramida o glucocerebrósido (GluCer) en el lisosoma, que, a su vez, es deacilada dando lugar a la glucosilesfingosina (GluSph o Liso-GL1) y, posteriormente, a esfingosina-1-fosfato (fig. 1), responsables, en parte, de la clínica y complicaciones de la enfermedad1,2.
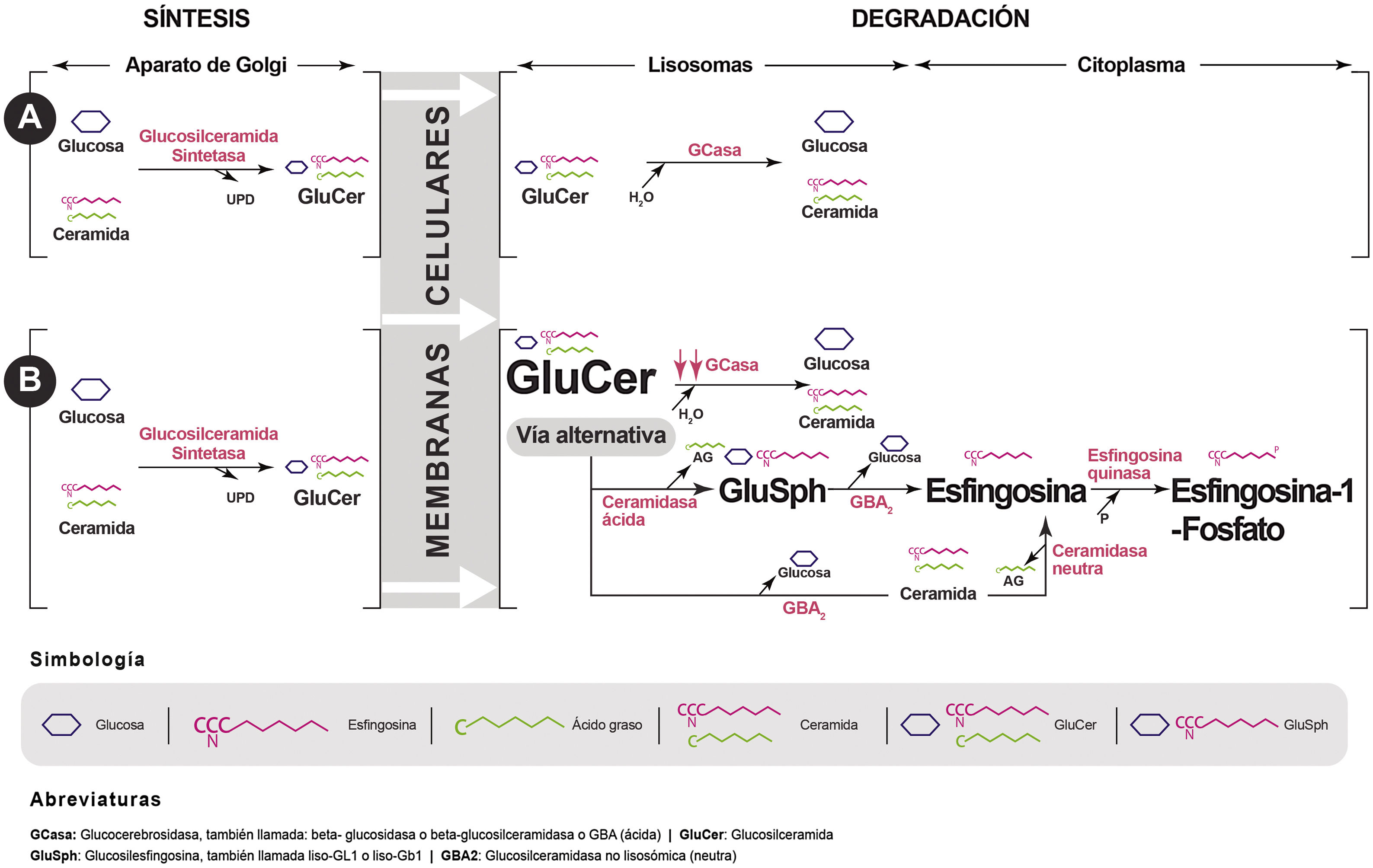
La variante tipo 1 (EG1) o no neuropática es la más frecuente en muestro medio, con una incidencia anual aproximada de un caso cada 50.000 recién nacidos vivos y con una prevalencia estimada de 1,5 casos/100.000 habitantes1,2. La EG1 se caracteriza por la aparición de esplenomegalia, trombocitopenia (u otra citopenia), hepatomegalia, dolor o necrosis ósea, deformación femoral distal, elevación de marcadores de inflamación (ferritina, fosfatasa ácida tartrato-resistente o enzima convertidora de angiotensina), hipergammaglobulinemia poli/monoclonal y hemorragias mucocutáneas en proporción variable1,2.
Desde hace unos 30 años, el tratamiento se ha basado en la administración del tratamiento de reemplazo enzimático (TRE), lo que ha supuesto una mejora espectacular en todos los aspectos relacionados con los pacientes. A principios de esta centuria se introdujo la terapia oral con inhibidores de la síntesis de sustrato (miglustat), que ha experimentado un cambio sustancial en los últimos años, al disponer de eliglustat. Este fármaco ha demostrado una eficacia similar a la del TRE y ha cambiado el manejo de estos pacientes. Sin embargo, todavía existe cierto grado de confusión en cuanto a las indicaciones y eficacia de los 2 fármacos inhibidores de la síntesis de GluCer disponibles actualmente: miglustat y eliglustat.
El objetivo principal de esta guía es actualizar y homogeneizar la situación actual del tratamiento oral de pacientes adultos con EG1 estableciendo las diferencias entre eliglustat y el tratamiento oral previamente existente, miglustat. Debido a que los 2 fármacos disponibles actúan inhibiendo a la glucosilceramida sintetasa (GCst), consideramos una necesidad aclarar las diferencias en cuanto a eficacia e indicación en el tratamiento de pacientes adultos con EG1. Esto incluye a pacientes, sin tratamiento previo, así como a previamente tratados con terapia enzimática, con o sin comorbilidades asociadas.
Material y métodosPara la elaboración del presente documento se creó un grupo de trabajo en EG en el adulto, compuesto por un total de 5 especialistas de Hematología, Hemoterapia y Medicina Interna de 5 hospitales españoles, con experiencia avalada en el diagnóstico y tratamiento de estos pacientes, así como en la elaboración de guías de práctica clínica.
En lo concerniente a los puntos de vista y preferencias de los pacientes, hemos de mencionar que no se ha contado con la participación directa de los mismos para la elaboración del presente documento, pero sí se ha utilizado información procedente de la literatura que aborda este tema.
Este documento va dirigido a facultativos de cualquier especialidad médica que deseen información sobre las indicaciones y el manejo del tratamiento oral de los pacientes con EG1, así como al resto de personal sanitario implicado en el mismo. En la elaboración se ha respondido a las cuestiones reflejadas en la tabla 1.
Preguntas que se han de responder en el tratamiento de reducción de sustrato en pacientes adultos con EG1
| 1. ¿Los tratamientos orales han demostrado eficacia, efectividad, eficiencia y seguridad? |
| 2. ¿Los tratamientos orales tienen la misma efectividad que el TRE? |
| 3. ¿Son eliglustat y miglustat igualmente equivalentes en eficacia, efectividad, eficiencia y seguridad? |
| 4. ¿Tienen eliglustat y miglustat la misma indicación? |
| 5. ¿Cuál es la dosis y la pauta de administración? |
| 6. ¿Pueden utilizarse en la edad infantil? |
| 7. ¿Pueden utilizarse en el embarazo y la lactancia? |
| 8. ¿Pueden utilizarse en pacientes polimedicados? |
| 9. ¿Pueden utilizarse en pacientes con cardiopatía, hepatopatía o nefropatía? |
| 10. ¿Está indicado eliglustat en pacientes con EG1 adultos previamente no tratados o pacientes tratados con TRE? |
| 11. ¿Está indicado el TRS en pacientes graves? |
| 12. ¿Es necesario estabilizar un paciente sin tratamiento previo con TRE antes de iniciar tratamiento con TRS? |
| 13. ¿Mejoran los biomarcadores de la enfermedad? |
| 14. ¿Son capaces de estabilizar, mejorar, revertir o prevenir las complicaciones óseas, hematológicas y/o viscerales de forma similar? |
| 15. ¿Eliglustat y/o miglustat mejora la calidad de vida? |
EG1: enfermedad de Gaucher tipo 1; TRE: tratamiento de reemplazo enzimático; TRS: tratamiento de reducción de sustrato.
La población diana a la que va destinado el documento son los pacientes varones o mujeres de 18 años o más, con EG1 que precisen iniciar tratamiento para su enfermedad o que prefieran o necesiten cambiar a tratamiento oral para su enfermedad. Las excepciones son pacientes con EG1: menores de 18 años, o embarazadas o que deseen ser madres próximamente, o varones que deseen tener descendencia próximamente, o aquellos cuyo genotipo metabolizador CYP2D6 contraindique el uso del tratamiento con inhibidores de la GCst, o que tengan comorbilidad o precisen fármacos que contraindiquen el uso de inhibidores de la GCst y cualquiera de los otros tipos de EG.
No se ha llevado a cabo en España ninguna prueba piloto sobre cualquiera de los tratamientos orales. En nuestro país existen aproximadamente 300 pacientes con EG1, de los cuales 39 llegaron a ser tratados con miglustat y un porcentaje significativo de los mismos debieron abandonarlo por sus efectos secundarios3. Desde 2017 hasta la fecha, se han tratado unos 100 pacientes con eliglustat sin que en España se haya documentado ningún dato de alarma o relacionado con la seguridad de los pacientes, según consulta llevada a cabo a través de la página web de la Agencia Española del Medicamento utilizando como palabra de búsqueda «eliglustat» (consulta realizada en junio de 2021: https://www.aemps.gob.es/tag/seguridad-8 y https://www.aemps.gob.es/acciones-informativas/notas-informativas-medicamentos-de-uso-humano).
Para alcanzar el objetivo, se decidió valorar los 2 medicamentos orales disponibles para el tratamiento de pacientes adultos con EG1 (miglustat y eliglustat) en cada una de las siguientes áreas: mecanismos de acción, evidencia de efectividad, indicaciones y contraindicaciones, seguimiento y tolerabilidad. La sistemática de trabajo que se siguió fue identificar, entre febrero y mayo de 2021 (mediante búsqueda en Embase, Cochrane y Medline), los artículos publicados acerca del tratamiento oral en la EG1, así como la ficha técnica de los 2 fármacos que se iban a analizar. Utilizando las palabras clave «eliglustat» y «miglustat» se identificaron 134 artículos en Medline, 16 en Cochrane y 32 en Embase.
La selección y clasificación posterior de los artículos que se iban a revisar se realizó según el nivel de evidencia: se consideraron en el nivel 1 las revisiones sistemáticas de ensayos clínicos (EECC) aleatorizados; en el nivel 2 los EECC aleatorizados bien diseñados; en el nivel 3 los EECC sin asignación aleatoria, estudios cohortes, de casos y controles y de series temporales con grupo control; y por último, en el nivel 4, las series de casos. Debido a la idiosincrasia de la EG1, clasificada como enfermedad ultrarrara, el grado de recomendación se categorizó en función del criterio clínico y la experiencia de los expertos. Con ello se alcanzaron las 61 citas que constan en la bibliografía del documento. Para las recomendaciones se utilizaron los grados de evidencia y niveles de recomendación de la Scottish Intercollegiate Guidelines Network (SIGN)4.
Durante las reuniones siguientes se presentó la evidencia seleccionada para, posteriormente, proceder a debatir las recomendaciones recogidas. A efectos de establecer el grado de consenso para cada una de las recomendaciones, los asistentes llevaron a cabo una votación, considerando como recomendaciones aceptadas aquellas en las que al menos 3 de los 5 autores se mostrasen de acuerdo.
Se invitó a 2 evaluadores externos, hematólogos/internistas expertos en EG1, para la revisión del rigor metodológico, mediante el instrumento Agree II. Las consideraciones de los revisores externos se utilizaron para enfatizar en los aspectos metodológicos y en la claridad de las recomendaciones.
Esta publicación ha recibido el aval científico de la Sociedad Española de Hematología y Hemoterapia (SEHH) y la Sociedad Española de Medicina Interna (SEMI).
ResultadosTratamiento de la enfermedad de Gaucher tipo 1Históricamente, la EG1 se ha tratado mediante cirugía tipo esplenectomía o procedimientos ortopédicos. Los nuevos tratamientos han cambiado de forma importante la historia natural de esta enfermedad, aunque sigue sin existir un tratamiento curativo5.
Los criterios para iniciar el tratamiento en los pacientes con EG1 deben valorarse de forma individualizada. Se suele recomendar iniciar el tratamiento en pacientes con enfermedad sintomática, incluyendo dolor –abdominal (visceromegalias) u óseo–, disfunción hepática, enfermedad pulmonar, fatiga, limitación al ejercicio, debilidad y caquexia, cualquier manifestación esquelética (incluyendo alteraciones radiológicas, como deformidad en matraz de Erlenmeyer), trombocitopenia (generalmente menos de 60.000 plaquetas/mm3, cifras en descenso o en aquellos casos con complicaciones hemorrágicas, independientemente del número de plaquetas), hemoglobina menor de 2g/dL del límite normal para su edad y sexo, y, en general, en todos los casos en que se objetive un deterioro de la calidad de vida o progresión de síntomas. En aquellos pacientes asintomáticos la indicación de tratamiento viene condicionada por la gravedad de la afectación ósea6,7.
Los objetivos generales a corto y largo plazo son7,8:
- 1)
Normalizar los niveles de hemoglobina, eliminando la dependencia transfusional.
- 2)
Mejorar la cifra de plaquetas con niveles por encima de las 100.000 plaquetas/mm3 y reducir la tendencia hemorrágica.
- 3)
Reducir la visceromegalia, prevenir la fibrosis hepática, cirrosis e hipertensión portal.
- 4)
Prevenir o mejorar la patología pulmonar (hipertensión pulmonar, síndrome hepatopulmonar).
- 5)
Prevenir las complicaciones óseas tales como: osteopenia, osteoporosis, necrosis avasculares, crisis óseas y las fracturas patológicas. Además de reducir o eliminar la analgesia crónica por dolores óseos.
- 6)
Mantener buena calidad de vida, promover un crecimiento óptimo, normalizar la expectativa de vida.
- 7)
En el embarazo, prevenir complicaciones durante el mismo, parto y posparto.
Actualmente, existen 2 tipos de tratamiento farmacológico aprobados para la EG1 (fig. 2). La primera modalidad es el TRE, cuya aprobación para pacientes con EG1 fue por primera vez a principios de los 90. El primer fármaco utilizado fue la alglucerasa (GCas placentaria purificada), que posteriormente fue reemplazado por la imiglucerasa. Más tarde fueron aprobadas velaglucerasa alfa en 2009 y taliglucerasa alfa en 20112,9,10. En nuestro país solo están disponibles comercialmente la imiglucerasa y la velaglucerasa alfa.
La dosificación inicial depende de la gravedad del paciente al diagnóstico: para pacientes graves o con enfermedad rápidamente progresiva y con comorbilidades, se empezará con 60U/kg cada 2 semanas; para pacientes graves o con enfermedad rápidamente progresiva, se empezará con 30-60U/kg cada 2 semanas, y para pacientes leves o con enfermedad moderada, con 15-30U/kg cada 2 semanas6,11. La dosis de mantenimiento se calcula tras al menos un año de la terapia inicial y se modifica según los resultados alcanzados6.
El TRE ha demostrado mejoría en todos los aspectos clínicos y parámetros biológicos de los pacientes con EG1. Los principales inconvenientes de esta terapia son la administración intravenosa, la dependencia del paciente del hospital, la posibilidad de desarrollo de aversión del paciente por las agujas y la necesidad de su infusión al inicio en medio hospitalario con el aumento del coste añadido10, si bien existe la posibilidad de que sea administrado si se cumplen las condiciones, en domicilio.
La segunda modalidad es el tratamiento de reducción de sustrato, aprobado a principios de 2000. El primer fármaco utilizado fue miglustat y, posteriormente, en 2015, se aprobó eliglustat. Ambos han demostrado efectos beneficiosos en la EG1, pero con importantes diferencias tanto en los mecanismos de acción como en los objetivos alcanzados, así como en los posibles efectos secundarios que uno u otro ocasionan y que quedarán explícitos en esta guía.
Existen actualmente otros tratamientos en desarrollo como las chaperonas o terapia génica, entre otros, además del tratamiento de soporte, que no vamos a abordar en esta guía.
Mecanismos de acciónEl tratamiento oral de la EG1 se basa en la utilización de fármacos reductores de sustrato, estrategia aplicada a las enfermedades lisosomales desde que fue descrita por primera vez por Radin et al. en 197212 y utilizada específicamente en la EG en 199613. Estos reducen la actividad de la GCst, que cataliza la síntesis de GluCer a partir de UDP-glucosa y ceramida. La inhibición de este primer paso en la producción de glucoesfingolípidos disminuye la producción de GluCer, que es el sustrato para la enzima deficiente en la EG. Además, permite que la mínima cantidad producida pueda ser degradada por la GCasa residual presente en todos los pacientes con EG1, eliminando los efectos patogénicos del acúmulo de GluCer y su principal derivado, la Liso-GL114 (fig. 1). Actualmente existen 2 fármacos inhibidores de la GCst, ambos con distinto mecanismo de acción y propiedades farmacológicas, lo que explica sus diferencias en la eficacia.
Miglustat es un derivado iminoazúcar (N-butildeoxinojirimicina) análogo a la D-glucosa, soluble en agua, que se desarrolló inicialmente como inhibidor de la α-glucosidasa, como tratamiento de la infección por el VIH. Posteriormente se observó que era un inhibidor reversible de la GCst en rango micromolar intermedio15. Su IC50 (concentración que produce el 50% de la inhibición) es de 20ul/L16.
Su principal desventaja reside en que es un inhibidor inespecífico de las α-glucosidasas i y ii, así como las disacaridasas intestinales (lactasa, sucrasa y maltasa), lo que condiciona la aparición de efectos adversos no deseables. Tras la administración de la dosis terapéutica de 100mg/8h, se absorbe, alcanzando pico máximo de concentración en plasma de 0,86ug/mL a las 2,5h, que disminuye discretamente si se administra con comida. Circula sin unirse a proteínas con un volumen de distribución de 83L.
Se elimina principalmente por vía renal tras una vida media de 6-7h, reduciéndose su eliminación en pacientes con insuficiencia renal. Aunque miglustat pasa la barrera hematoencefálica (PM 219,28g/mol, soluble en agua) carece de efecto sobre la clínica neurológica de los pacientes con EG demostrable en ensayos clínicos16.
El tartrato de eliglustat (Genz-112638) es un análogo de ceramida con estructura similar al D-treo-1-fenil-2-decanoilamino3-morfolino-propanol. Su actividad inhibitoria es reversible, potente y selectiva de la GCst con una IC50 en rango nanomolar (10ng/mL, 24nM)14,17–19, sin inhibir la función de las disacaridasas intestinales. Eliglustat tiene una biodisponibilidad oral baja (<5%) porque experimenta un importante efecto de primer-paso hepático al ser metabolizado por el citocromo hepático CYP2D6 y en menor grado por el CYP3A. Este hecho condiciona su farmacocinética según el fenotipo metabolizador de los pacientes para CYP2D6. Así, para aquellos metabolizadores rápidos (MR) o intermedios (MI), eliglustat tiene una farmacocinética no lineal, dependiente de tiempo, y mayor que la farmacocinética dosis-dependiente; por el contrario, los metabolizadores lentos (ML) siguen una farmacocinética lineal y tiempo-independiente. La dosis terapéutica se ajustará según el fenotipo metabolizador de los pacientes.
Tras la administración de la dosis terapéutica, se absorbe, alcanzando pico máximo de concentración en plasma de 12,1-25ng/mL (MR) o 113-137ng/mL (ML) a las 1,5h (MR) o 3h (ML)17,18. Su absorción no varía con las comidas, aunque disminuye discretamente tras comida rica en grasas. Circula en plasma unido a proteínas (76-83%). Su volumen de distribución es muy elevado (835L) lo que asegura su amplia distribución tisular.
Se elimina por heces (51%) y orina (42%), tras una vida media variable de 6,5h (MR) a 8,9h (ML), dependiendo del grado de metabolización17–19. Eliglustat, a pesar de su bajo peso molecular (PM 404,5g/mL), no atraviesa la barrera hematoencefálica al ser un sustrato de la P-glucoproteína14,17.
EvidenciaMiglustatSegún los datos de los ensayos clínicos20–22, miglustat fue aprobado en la Unión Europea en 2002 y en EE. UU. en 2003. Los estudios realizados en adultos con EG1 sin tratamiento previo y con formas leves o moderadas demostraron unos resultados efectivos en la reducción de los volúmenes de bazo y de hígado y en un incremento de las cifras de hemoglobina y plaquetas, durante los 12-36 meses de tratamiento23,24. También demostró una mejoría en la densidad mineral ósea tanto a nivel trabecular como cortical de los huesos25, pero, como los pacientes incluidos en los estudios son leves o moderados y, por lo tanto, con poca afectación ósea, no es posible sacar conclusiones de la respuesta clínica ósea con este tratamiento. Por ello, se concluyó que miglustat podía servir como terapia de mantenimiento en algunos pacientes previamente estabilizados con TRE que, por alguna razón, ya no podían tomarlo.
Sin embargo, estudios posteriores mostraron que un grupo de pacientes a los que se les había cambiado a miglustat después de estabilizarse con TRE mostraban un aumento de quitotriosidasa y disminución de hemoglobina y plaquetas, lo que indicaba una progresión de la enfermedad20,24,26, y, a partir del segundo año de tratamiento en monoterapia con miglustat, el 26% de los pacientes lo abandonaba por eventos adversos o por progresión de la enfermedad3.
No obstante, los principales problemas del fármaco son los efectos secundarios, especialmente los gastrointestinales, como diarrea y flatulencia, que son la principal causa para abandonar el tratamiento25. Otros efectos secundarios importantes son pérdida de peso, temblor y neuropatía periférica, así como los trastornos neurológicos que afectan hasta al 29% de los pacientes27. Todo esto ha hecho que el abandono del tratamiento sea muy alto y, aunque varía mucho según los países (en España fue del 13% mientras que en Francia y Reino Unido fue del 68 y 69%, respectivamente), casi el 50% de los pacientes dejó el tratamiento3.
Por lo tanto, miglustat es un iminoazúcar indicado como tratamiento de segunda línea en aquellos pacientes con EG1 leve o moderada que por distintas razones no pueden ser tratados con TRE o con eliglustat. No se aconseja que ningún paciente de nuevo diagnóstico inicie terapia con miglustat28 salvo los pocos casos con EG1 que toleraron bien el tratamiento y que han continuado con él desde hace años3.
EliglustatLa metodología de búsqueda empleada permitió la selección de los EECC controlados que a continuación se detallan.
Estudios de eficaciaEnsayos clínicos fase ii (NCT00358150)El ensayo fundamental para evaluar la eficacia de eliglustat en la EG1 se llevó a cabo entre 2010 y 2019 en 26 adultos no tratados previamente. En los 19 pacientes que finalizaron el seguimiento a los 8 años se halló una importante mejoría en las visceromegalias (con reducción del bazo y el hígado un 69% y un 34%, respectivamente); en los parámetros hematológicos (con incremento de la hemoglobina 2,2g/dL y las plaquetas un 113%); en los biomarcadores clásicos y Liso-GL1, así como una mejoría en la calidad de vida y se objetivó una normalización de los T-score lumbares que previamente mostraban osteopenia29–32.
Ensayos clínicos fase IIISe han publicado 3 ensayos fundamentales, aleatorizados, multicéntricos y multinacionales:
ENGAGE (NCT00891202)- -
Ensayo doble ciego y controlado con placebo en 40 pacientes con EG1 sin tratamiento previo, que fueron aleatorizados 1:1 a tratamiento con eliglustat o placebo durante 9 meses. Todos los pacientes tenían esplenomegalia con trombocitopenia y/o anemia. El objetivo principal de valoración fue el cambio porcentual del volumen esplénico; como criterios de eficacia secundarios, se evaluaron los parámetros hematológicos, y, como criterios terciarios, se establecieron la mejoría de biomarcadores y de los parámetros óseos.
A los 9 meses se demostró mejoría significativa en los recuentos de hemoglobina (+0,69g/dL vs. −0,54g/dL) y plaquetas (+32% vs. −9%) y del tamaño del hígado (−5,2% vs.+1,4%) y el bazo (−27,8% vs.+2,3%) que disminuyeron con eliglustat, pero no con el placebo. En el grupo eliglustat también se produjo mejoría estadísticamente significativa del daño medular óseo, así como de los biomarcadores (p=0,0021), y no con el placebo33. En una segunda extensión, la continuación del tratamiento con eliglustat durante 9 meses más resultó en una mejora significativa de todos los parámetros de la enfermedad y los que previamente habían recibido placebo también experimentaron mejoría significativa de los volúmenes del bazo y del hígado, hemoglobina y plaquetas. El tratamiento con eliglustat también se asoció con una mejora en la puntuación del daño medular óseo, la densidad mineral ósea y los biomarcadores34.
- -
Evaluó la eficacia y seguridad de eliglustat en términos de no inferioridad en pacientes estabilizados con imiglucerasa. Entre los 106 de un total de 159 pacientes con EG1 que cambiaron a eliglustat o mantuvieron la enzima durante 12 meses, todas las características clínicas y los parámetros hematológicos fueron estables en ambos grupos. Asimismo, el daño medular óseo no se modificó en este período de tiempo35.
En la extensión de este estudio a largo plazo, después de un año, todos los pacientes continuaron o cambiaron a eliglustat, demostrándose que el tamaño del hígado y el bazo, así como los recuentos de hemoglobina y plaquetas, se mantuvieron estables durante 1-4 años de tratamiento. En esta extensión también se demostraron incrementos significativos de los Z-score lumbar (p<0,0001 durante los 4 años) y femoral (p=0,005 al primer año y p=0,02 al tercero)36.
- -
Ensayo doble ciego y de comparación de la dosificación de eliglustat una vez al día, con el régimen aprobado de 2 veces al día, en adultos con EG1 durante una mediana de 3,3 años. Se demostró que los pacientes con eliglustat 2 veces al día mostraron una mayor estabilidad en general37.
Existen muy pocas publicaciones de eliglustat fuera de los ensayos clínicos que tengan suficientes casos como para sacar conclusiones.
Mistry et al. publicaron en 202038 un estudio de 231 pacientes (89% de EE. UU.) con EG1 del Registro Internacional y que dividen en 3 grupos: 19 pacientes nuevos, 212 pacientes que cambian de TRE no esplenectomizados y 36 pacientes que cambian de TRE esplenectomizados. Todos ellos habían estado con eliglustat una media de 2 años (mínimo un año) obteniendo resultados al inicio y al final del estudio. En todos los pacientes nuevos se observa una mejoría clínica y estadísticamente significativa, tanto de los parámetros hematológicos como en las organomegalias, y una estabilización o una pequeña mejoría de los pacientes que cambiaron de la TRE, estando todos ellos dentro de los objetivos terapéuticos que se establecieron para el TRE.
Los valores de quitotriosidasa también disminuyeron significativamente lo que indica una regresión de la inflamación y de la polarización de los macrófagos aberrantes que subyace en la fisiopatología de esta enfermedad. La valoración ósea fue limitada por el corto período de tiempo del estudio, pero en los pacientes no esplenectomizados que cambiaron del TRE se vio un incremento significativo del Z-score de la columna lumbar después del tratamiento con eliglustat.
Estos datos corroboran los resultados obtenidos en los ensayos clínicos pivotales. Los mejores resultados se obtuvieron en los pacientes que tenían peores datos al inicio del tratamiento. Eliglustat fue muy bien tolerado y discontinuaron el tratamiento un 9% (22/231) de los pacientes por diferentes razones, tales como efectos adversos, preferencia por el tratamiento enzimático, falta de disponibilidad del fármaco o por no cubrirlo su seguro médico, y, en 7 casos, por la intención de quedarse embarazada.
Evidencia más limitada encontramos en un estudio retrospectivo multicéntrico de 14 pacientes en tratamiento con eliglustatEl comportamiento de los biomarcadores Liso-GL1 y quitotriosidasa en los pacientes tratados con eliglustat frente a los tratados con las enzimas actualmente disponibles en el mercado, con un mejor comportamiento en la reducción de los biomarcadores asociados a la EG, es estadísticamente significativo en aquellos pacientes del grupo de eliglustat39.
Estudios sobre toxicidad, farmacocinética o efectos secundarios- -
Análisis agrupado de eventos adversos de Peterschmitt: entre 393 pacientes con EG1 tratados con eliglustat en los 4 ensayos clínicos (NCT00358150, NCT00891202, NCT00943111 y NCT01074944), no hubo eventos adversos graves o interrupciones del fármaco por elevaciones de ALT o lesión hepática aguda40. En el estudio de seguimiento llevado a cabo por los mismos autores se evaluó la toxicidad o efectos secundarios por eliglustat en los 4 trabajos mencionados, representando una exposición de 1400 pacientes-año al mencionado fármaco y una duración media del tratamiento de 3,6 años (máximo: 9,3 años): el 81% de los pacientes permanecieron en su ensayo respectivo hasta la disponibilidad comercial de eliglustat o hasta la finalización del ensayo; 9 pacientes (2,3%) se retiraron debido a uno o más eventos adversos informados como relacionados con eliglustat y todos, excepto uno, fueron leves o moderados. En general, el 97% de los eventos adversos fueron leves o moderados y el investigador informó que el 86% no estaban relacionados con eliglustat. La tasa general de eventos adversos disminuyó con el tiempo y no se correlacionó con la dosis del fármaco41.
- -
Impacto del daño hepático y renal sobre la tolerabilidad y farmacocinética en pacientes con EG1 tratados con eliglustat: analiza los datos de los estudios de fase i (NCT02536937 / NCT02536911) después de una dosis única de 84mg de eliglustat. Los pacientes MR con insuficiencia hepática (IH) moderada e insuficiencia renal grave presentaban mayores concentraciones en plasma de eliglustat. También se predijeron exposiciones más altas de eliglustat en MR con IH leve después de la coadministración con un inhibidor de CYP2D6 o CYP3A a dosis repetidas42.
- -
Efecto de eliglustat sobre la farmacocinética de digoxina (sustrato de la glucoproteína-P), metoprolol (sustrato del CYP2D6) y anticonceptivos orales (sustrato del CYP3A) y la absorción de eliglustat cuando se coadministra con inhibidores de la secreción gástrica: se inscribieron sujetos sanos en 4 estudios clínicos de fase i, comprobándose que eliglustat fue bien tolerado y, cuando se coadministra con medicamentos que son sustrato de la glucoproteína P o CYP2D6, se requieren dosis más bajas de estos. Por otro lado, eliglustat puede coadministrarse con anticonceptivos orales e inhibidores de la secreción gástrica sin modificaciones de dosis43.
Además de lo ya comentado en lo concerniente a los ensayos ENGAGE y ENCORE, existe una publicación específicamente centrada en la valoración ósea:
- -
Los efectos esqueléticos de eliglustat fueron evaluados mediante la monitorización prospectiva de la densidad mineral ósea, el desarrollo de fracturas, la infiltración medular por células de Gaucher, la aparición de lesiones óseas focales e infartos durante el ensayo de fase ii NCT00358150. El estudio demostró que la densidad mineral ósea de la columna lumbar aumentó significativamente (p=0,02; n=15) en una media del 9,9% (14,2%) desde el inicio hasta el año 4. Los T-scores correspondientes a la columna lumbar aumentaron significativamente (p=0,01) desde una media de −1,6 (1,1) a −0,9 (1,3). Los T-scores del fémur se mantuvieron normales durante 4 años. La resonancia magnética (RM) del fémur mostró que 10/18 (56%) pacientes tenían menor infiltración medular en comparación con las imágenes iniciales y un paciente con mejoría temprana ósea tuvo un empeoramiento transitorio en el año 4. No se describieron fracturas o crisis óseas.
Al inicio del estudio, 8/19 (42%) pacientes tenían lesiones óseas focales, que permanecieron estables, y 7/19 (37%) pacientes tuvieron infartos óseos, que mejoraron en un paciente al año 2. En el año 4, se descubrió una lesión ósea previamente inexistente, asintomática e indeterminada, que posteriormente se resolvió31.
Análisis post-hocAparte del ENCORE36 otros 2 análisis post-hoc compararon la estabilidad clínica entre el uso de imiglucerasa o velaglucerasa y eliglustat.
- -
Ibrahim et al. compararon la respuesta a eliglustat en pacientes con EG1 no tratados previamente en los ensayos clínicos fase ii y iii (ENGAGE) con la respuesta a imiglucerasa en otra cohorte de pacientes sin tratamiento previo y pertenecientes al Registro Internacional Colaborativo de Gaucher: los parámetros viscerales y hematológicos mejoraron significativamente en ambos grupos de un modo similar sin que se incluyeran los efectos sobre el hueso o se tomaran datos sobre posibles efectos adversos44.
- -
Pleat et al. llevaron a cabo un subanálisis del estudio ENCORE en el que compararon la estabilidad en pacientes adultos con EG1 que cambiaron de velaglucerasa a imiglucerasa o a eliglustat: el 88% de los pacientes que cambiaron a imiglucerasa y el 90% de los que lo hicieron a eliglustat alcanzaron los objetivos del ensayo45.
Existe un único estudio con el objetivo de comparar la eficacia de imiglucerasa con eliglustat en el tratamiento de pacientes con EG. En el mismo se realizaron búsquedas en PubMed / Medline, Cochrane Library, Scopus, Web of Science, Embase y Google Scholar hasta agosto de 2018, incluyendo todos los estudios aleatorizados, controlados cuasialeatorios y de cohortes sobre pacientes con EG1 en los que se comparó imiglucerasa con eliglustat.
Los resultados mostraron que no existe una diferencia significativa entre los 2 medicamentos en términos de aumento de la hemoglobina en sangre, recuento de plaquetas y reducción del tamaño del hígado y el bazo. Los hallazgos de esta revisión mostraron que ambos medicamentos son efectivos en el tratamiento de la EG1 y no hay diferencias estadísticamente significativas entre sus eficacias46.
Estudio sobre el impacto económicoNalysnyk et al. llevaron a cabo un estudio sobre el impacto económico en el sistema sanitario de EE. UU. asociado con la transición de pacientes que reciben TRE a eliglustat para el tratamiento de adultos con EG1. Los costes anuales de TRE se calcularon asumiendo una dosis quincenal de 47,4U/kg; por ejemplo, en un paciente de 72kg de peso y 24 infusiones por año. Los resultados del estudio demostraron que un mayor uso de eliglustat administrado por vía oral –en lugar del más caro basado en la infusión enzimática– significaba un ahorro significativo47.
Indicaciones / contraindicacionesLos 2 fármacos inhibidores orales de la GCst, miglustat y eliglustat, tienen distinta farmacocinética y efectividad, lo que origina indicaciones y precauciones claramente distintas.
Miglustat está indicado para pacientes adultos con EG1 leve o moderada, en los que no sea adecuado el TRE. El criterio de enfermedad leve o moderada es arbitrario, pero incluiría a pacientes con hemoglobina mayor de 9g/dL, plaquetas superiores a 50×109/L y sin evidencia de enfermedad ósea progresiva15. No aporta ninguna ventaja sobre el TRE, siendo menos efectivo que este28 y teniendo como única ventaja la de ser un fármaco de administración oral23. A pesar de que se haya valorado la administración concomitante con TRE en pacientes con respuesta insuficiente a máximas dosis15, esta pauta de administración no se debe utilizar fuera de ensayos clínicos28. La única contraindicación absoluta es la hipersensibilidad al fármaco o alguno de sus componentes.
No se ha evaluado en pacientes con IH. En pacientes con insuficiencia renal es preciso reducir la dosis según el filtrado glomerular (FG) (100mg/12h si FG 50-70mL/min/1,73m2, y 100mg/día si FG 30-50mL/min/1,73m2) y se desaconseja su uso cuando el FG sea inferior a 30mL/min/1,73m2.
No se ha establecido su efectividad en niños o adolescentes menores de 17 años, ni hay experiencia en mayores de 70 años. Atraviesa la barrera placentaria y se excreta en leche materna por lo que no debe emplearse durante el embarazo o la lactancia. Las mujeres en edad fértil deberán utilizar sistemas anticonceptivos. Dado que afecta a los parámetros espermáticos, está contraindicado en varones que deseen concebir15,28,48.
Eliglustat está indicado para pacientes adultos con EG1 que son ML, MI o MR, sin poder utilizarse en los ultrarrápidos (MUR)49.
Está contraindicado en casos de hipersensibilidad al fármaco o a sus componentes49. Igualmente se contraindica en pacientes MI y MR de CYP2D6 que tomen un inhibidor potente o moderado de dicho citocromo, asociado a otro inhibidor potente o moderado del CYP3A, así como en pacientes ML del CYP2D6 que tomen un inhibidor potente de CYP3A49,50. Por su metabolismo hepático, está contraindicado en pacientes MR con IH grave (Child-Pugh clase C), o bien si es leve/moderada (clase A o B) asociada a la toma de un inhibidor potente/moderado de CYP2D649.
Se detalla más información sobre la combinación de eliglustat con otros fármacos y sustancias (pomelo, etc.) en el apartado Tolerabilidad. Dado que eliglustat utiliza lactosa como excipiente, los pacientes con intolerancia hereditaria a la galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa, no deben tomarlo49.
En pacientes con IH, aparte de las contraindicaciones anteriormente mencionadas, eliglustat no se recomienda en MR de CYP2D6 con IH moderada (Child-Pugh clase B), ni en MI o ML con cualquier grado de IH. En pacientes MR con IH leve (clase A) que tomen un inhibidor leve del CYP2D6 o cualquier inhibidor de CYP3A, se recomienda reducir la dosis a una toma de 84mg/día49,51.
Un estudio reciente fase i en población sin EG1 ha demostrado que en MR con IH moderada, las concentraciones séricas de eliglustat, así como su Cmáx, AUC y t1/2 son mayores que en aquellos con enfermedad leve o sin afectación hepática42. Los modelos farmacocinéticos predicen mayor exposición al fármaco en los MR con IH leve y, sobre todo, con afectación moderada, comparada con controles sanos, y estos niveles se incrementan al asociar inhibidores de CYP2D6 y CYP342. El trabajo concluye que la afectación hepática leve, por sí sola, no tiene efecto sustancial sobre la farmacocinética de eliglustat en dosis única en MR, pero sí que se detecta dicho efecto cuando la IH es moderada42.
En pacientes MR con insuficiencia renal se puede utilizar eliglustat sin modificar la dosis. El mismo estudio fase i en población sin EG142 concluye que la exposición a eliglustat en sujetos MR con insuficiencia renal grave es similar a población sana. Para aquellos MI o ML con cualquier grado de afectación renal, o bien MR con insuficiencia en grado terminal, no se recomienda su uso49,51.
En pacientes con afectación cardíaca tipo insuficiencia cardíaca congestiva, infarto agudo de miocardio, bradicardia, bloqueo cardíaco, arritmia ventricular y síndrome de QT largo, debería evitarse el uso de eliglustat ya que puede aumentar el intervalo QT cuando sus concentraciones plasmáticas son sustancialmente elevadas (11 veces la dosis terapéutica)49–51. Por esta razón y debido a la interacción entre fármacos, debería evitarse la combinación de eliglustat con antiarrítmicos que alarguen el intervalo QT como los de clase IA clase iii49,50 o clase IC51.
Datos in vitro han demostrado que eliglustat inhibe mínimamente al canal de potasio hERG y los canales de sodio Nav1.5 y Calcio Cav1.252. Sin embargo, los ensayos realizados en población sana (fase i) utilizando dosis supraterapéuticas (800mg) no han demostrado cambios electrocardiográficos significativos en el intervalo QT, QRS o PR52. Ya que se ha demostrado que existe correlación lineal entre la concentración de eliglustat y los cambios en QT, se pueden predecir alteraciones significativas al administrar un inhibidor potente de CYP2D6 y otro similar de CYP3A de forma conjunta (combinación contraindicada en ficha técnica)52.
En la práctica clínica, no se han detectado casos de pacientes con prolongación clínica significativa del intervalo QT, cambios en la repolarización, arritmia torsade de pointes o ventricular mantenida, ni bloqueo AV de segundo grado mayor de tipo 1 o cualquier otro signo de toxicidad cardiaca, tanto en los ensayos fase ii y iii (ENGAGE, ENCORE y EDGE) como desde la comercialización de eliglustat51,52. Por todo lo aquí expuesto se recomienda estudio cardiológico previo al inicio del tratamiento con eliglustat50.
La experiencia de eliglustat en mayores de 65 años es limitada, pero no parece haber diferencias con los menores, ni se precisa ajuste de dosis, aunque hay que prestar especial atención a la medicación concomitante49,50. No se ha establecido la eficacia en niños o menores de 18 años, por lo que, hasta obtener resultados del ensayo clínico específico (ELIKIDS, NCT03485677), no se recomienda su uso en estos pacientes49,50. Los datos de eliglustat en gestantes son escasos y proceden de los EECC fase ii y fase iii (ENGAGE, ENCORE y EDGE) y fase i (población sana)53, considerándose categoría C51. De entre las 202 mujeres incluidas en estos 4 EECC, 18 mujeres tratadas tuvieron 19 gestaciones, con una mediana de exposición a eliglustat de 38 días. Trece gestaciones llegaron a término, dando lugar a 14 niños sanos, con solo un aborto espontáneo y una muerte intraútero en la misma paciente, no relacionados con eliglustat. De las voluntarias incluidas en el fase i, se produjo un aborto espontáneo en una mujer con antecedentes de aborto previo53. Aunque esta tasa de aborto espontáneo (5%) es inferior al registrado en la población general (12-24%), de forma preventiva, se debería evitar el uso de eliglustat durante el embarazo y cambiar a TRE49–51.
Los datos obtenidos de modelos animales demuestran que eliglustat se secreta en leche materna, por lo que deberá ponderarse el beneficio materno frente al del lactante49–51. Se ha observado en animales que eliglustat a altas dosis puede afectar a la espermatogénesis de forma reversible49,50. Sin embargo, los datos obtenidos de los 4 EECC fase ii y i han demostrado que es posible la gestación en mujeres parejas de hombres tratados con eliglustat. De entre los 393 pacientes incluidos en estos 4 EECC, 16 varones tratados han sido padres de 18 niños sanos53.
En pacientes que vayan a ser sometidos a cirugía, es recomendable suspender temporalmente la toma de eliglustat50.
En resumen, miglustat es un tratamiento de segunda línea indicado para el reducido grupo de pacientes con EG1 en los que el tratamiento de primera línea no sea posible por motivos médicos y de acceso o cumplimiento terapéutico15,23,28,48. Por contra, eliglustat es un tratamiento de primera línea, similar a la TRE, aplicable a todos los pacientes con EG1 que no sean MUR, siempre que la medicación concomitante o la IH grave del paciente no lo contraindiquen49–51.
SeguimientoEl seguimiento de los pacientes tratados con eliglustat varía poco en comparación con aquellos que reciben el TRE7,54,55. Previo al inicio del tratamiento se deberá haber llevado una evaluación cardiológica, cuantificando el fenotipo a través de los índices de gravedad de Zimram56, GAUSSI-157 y GD-DS358, así como mediante el uso de los biomarcadores, y una RM de esqueleto con densitometría. Todos los pacientes deberán someterse a una evaluación regular completa, cuya frecuencia depende de si se han alcanzado los objetivos terapéuticos de la siguiente manera:
- •
Hasta conseguir los objetivos:
- ∘
Cada 3 meses: evaluación hematológica y biomarcadores.
- ∘
A los 12 meses: exploración física, encuesta SF-36, TC o RM de hígado y bazo, RM de esqueleto y densitometría ósea.
- •
Una vez conseguidos los objetivos:
- ∘
Anualmente: exploración física y encuesta SF-36.
- ∘
Anual o bianualmente (individualizando): evaluación hematológica, biomarcadores, TC o RM de hígado y bazo, RM de esqueleto y densitometría ósea.
Además, se debe realizar una reevaluación completa e inmediata en caso de que se produzca una complicación clínica significativa.
Por lo que respecta a los biomarcadores, en los estudios en fase ii, todos demostraron una disminución muy significativa con eliglustat ya desde el primer año, estabilizándose o ligeros descensos, a partir del cuarto. Liso-GL1 mostró un descenso del 60% en el primer año, llegando al 80% a partir del segundo y estabilizándose el descenso en un 92% del resultado que tenían al diagnóstico, en el octavo año31. Lo que nunca se debe esperar, ni con eliglustat ni con ningún otro tratamiento, es conseguir una cifra normal de ningún biomarcador, por lo que esto no debe ser un objetivo terapéutico7,54.
TolerabilidadComo ha quedado reflejado ya en esta guía de tratamiento oral de la EG1, aunque disponemos de 2 fármacos aprobados para su uso oral en esta patología (miglustat y eliglustat), ambos son muy diferentes en cuanto a su eficacia y tolerabilidad. Por lo tanto, como tratamiento de primera línea, nos referiremos a las cuestiones específicas de tolerabilidad de eliglustat.
Eliglustat y genotipo de CYP2D6Eliglustat es un fármaco que se metaboliza a nivel hepático por el citocromo CYP2D6 y en menor medida por CYP3A4. En la tabla 2 recogemos algunas variantes genéticas de CYP2D6 con su correspondiente actividad metabólica59.
Por ello, antes de indicar tratamiento con eliglustat a un paciente debemos conocer su fenotipo metabólico respecto a CYP2D649. Este paso es crítico en el inicio y establecimiento del tratamiento, puesto que en función de dicho fenotipo metabólico sabremos: (1) si el paciente puede recibir o no el tratamiento, y (2) la dosis adecuada para cada paciente. Como ya se ha comentado en el apartado Mecanismos de acción, la indicación de tratamiento según el fenotipo y la dosis del fármaco se recogen en la tabla 359,60.
La obligatoriedad de determinar el fenotipo metabólico de un paciente que va a recibir eliglustat, lejos de ser un condicionante negativo, nos acerca al uso de la Farmacogenética como herramienta de ayuda en la denominada Medicina Personalizada o de Precisión60. De esta forma sabremos de antemano no solo qué pacientes se beneficiarán del fármaco y quiénes no, si no que podremos determinar cuál es la dosis adecuada para conseguir los objetivos terapéuticos y cómo modificarla en función de la administración de otros fármacos que compartan la misma vía metabólica.
Interacciones farmacológicasLos fármacos metabolizados a través de los citocromos CYP2D6 y CYP3A4 pueden comportarse como simples sustratos de la enzima, inductores o inhibidores de las mismas. Así pues, nos encontraremos con fármacos que pueden alterar la capacidad metabólica de dichos citocromos, modificando la concentración final de eliglustat disponible tras una dosis dada, siendo el grado de interacción variable en función del fenotipo metabólico del paciente.
En general, podemos decir que el uso de eliglustat junto a un inhibidor potente de CYP2D6 o CYP3A4 haría que el fármaco se degradara más lentamente, acumulándose el fármaco activo y disminuyendo sus metabolitos inactivos y podría exponer al paciente a soportar una mayor toxicidad del mismo. Por otra parte, el uso de eliglustat con inductores potentes de CYP3A4 haría que el fármaco se degradara de una manera mayor a lo establecido de forma habitual, traduciéndose en la clínica este efecto en una disminución o ausencia del efecto terapéutico deseado60.
A modo de cuadro resumen, en la tabla 4 podemos establecer las indicaciones de uso y la dosis de eliglustat requerida cuando se usa de forma concomitante con fármacos que comparten ruta metabólica50,51,60,61.
Dosis de eliglustat según el uso de otros posibles fármacos concomitantes
| Fármaco asociado | Uso / Dosis recomendada | ||
|---|---|---|---|
| Metabolizador rápido | Metabolizador intermedio | Metabolizador lento | |
| Inhibidor fuerte CYP2D6 (p. ej., paroxetina, fluoxetina, quinidina, bupropion) | No recomendado | No recomendado | Sin datos. Evitar |
| Inhibidor moderado CYP2D6 (p. ej., duloxetina, terbinafina, moclobemida, mirabegron, cinacalcet, dronedarona) | 84mg/24h | 84mg/24h | Sin datos. Evitar |
| Inhibidor fuerte CYP3A (p. ej., ketoconazol, claritromicina, itraconazo, cobicistat, indinavir, lopinavir, ritonavir, saquinavir, telaprevir, tipranavir, posaconazol, voriconazol, conivaptan, boceprevir) | 84mg/24h | Contraindicado | Contraindicado |
| Inhibidor moderado CYP3A (p. ej., fluconazol, eritromicina, ciprofloxacino, diltiazem, verapamilo, aprepitant, atazanavir, darunavir, fosamprenavir, imatinib, cimetidina) | 84mg/24h | 84mg/24h | No recomendado |
| Inhibidor (F o M) CYP2D6+CYP3A | Contraindicado | Contraindicado | Contraindicado |
| Inductor CYP3A (p. ej., rifampicina, rifampin, rifabutin, fenobarbital, carbamazepina, fenitoína, dexametasona, hierba de San Juan) | No recomendado | No recomendado | No recomendado |
De manera recíproca, el uso de eliglustat puede condicionar la concentración de otros fármacos prescritos en el mismo paciente y que usen tanto la vía de CYP2D6 como la de la glucoproteína P. Así, ante la necesidad de utilizar de forma eventual alguno de estos fármacos en un paciente con EG1 que toma eliglustat, se debe considerar un potencial ajuste de su dosis.
Presentamos en la tabla 5 algunos de los ejemplos de fármacos que usan la vía de CYP2D6 y la vía de glucoproteína P durante su metabolismo.
Fármacos metabolizados mediante CYP2D6 y la vía de GP-P
| Fármacos metabolizados como sustratos vía CYP2D6 | |
|---|---|
| Anfetaminas | Anfetamina, dexfenfluramina, metoxianfetamina |
| Antiarrítmicos | Lidocaína, encainida, flecainida, mexiletina, propafenona, quinidina |
| Antidepresivos | Amitriptilina, clomipramina, imipramina, desipramina, nortriptilina, paroxetina, atomoxetina, venlafaxina |
| Antipsicóticos | Clorpromazina, clozapina, haloperidol, risperidona, tioridazina, zuclopentisol, quetiapina, aripiprazol, ziprasidona |
| β-bloqueantes | Alprenolol, carvediol, labetalol, metoprolol, propranolol, timolol |
| Opiáceos | Codeína, etilmorfina, hidroxicodona, tramadol, dextrometorfano |
| Otros | Clorpropamida, flunarizina, tamoxifeno, ondansetrón, tropisetrón, clorfeniramina, loratadina |
| Fármacos que tienen paso por GP-P | |
| Digoxina, dabigatran, colchicina, tacrolimus, quinidina, dexametasona | |
GP-P: glucoproteína P.
Se pueden consultar otros recursos digitales para fármacos no descritos, entre otros: www.uptodate.com, www.drugs.com y https://reference.medscape.com/drug-interactionchecker.
Efectos adversosLos efectos secundarios documentados más frecuentemente como los relacionados con el tratamiento fueron: dispepsia, cefalea, dolor abdominal e inestabilidad. Se han informado en el 5-6% del total de pacientes estudiados. La mayor parte de estos eventos fueron leves y se documentaron solo en una ocasión en la mayoría de los pacientes. Además, a medida que aumenta el tiempo en que el paciente está en tratamiento con eliglustat se comunican menos efectos adversos30,32–34.
No existe diferencia en la aparición de efectos secundarios en los diferentes fenotipos metabólicos (metabolizadores rápidos, intermedios o lentos) lo cual demuestra la eficacia que tiene la determinación previa del genotipo de CYP2D6 en los pacientes que van a recibir el fármaco.
Cuando se comparan los efectos adversos asociados a eliglustat con los relacionados con miglustat, se observa que no hay efecto de clase en el tratamiento de reducción de sustrato. La toxicidad documentada con miglustat (diarrea [80%], pérdida de peso [65%] y temblor [30%]) apenas aparece con la toma de eliglustat (14; 0,8 y 0,8% respectivamente).
DiscusiónEn el presente artículo se recogen las recomendaciones de este grupo de trabajo en relación con el tratamiento oral de la EG1 del adulto, siempre teniendo en cuenta que cada paciente debe ser valorado de forma individual y sus circunstancias particulares. Si bien la principal limitación de este trabajo viene dada de la escasa evidencia encontrada, secundaria a la baja prevalencia de la EG1, tiene la utilidad de ser la primera guía que compara y establece distintas recomendaciones para el uso de los diferentes fármacos orales en EG1. Nuestra intención es valorar regularmente las evidencias publicadas en este campo por si fueran necesarias modificaciones del documento. Si se detectaran nuevas aportaciones científicas que entraran en conflicto con lo descrito en esta guía, se procederá a realizar una nueva edición.
Las presentes recomendaciones tendrán un período de vigencia de 2 años desde esta primera publicación y se prevé una actualización mediante una revisión sistemática bibliográfica y por revisores externos en noviembre de 2023. Los encargados de llevar a cabo la mencionada actualización serán los autores de este manuscrito.
Por las características de este documento de consenso no se puede evaluar la aplicabilidad o su utilidad.
ConclusionesSe pueden resumir en los siguientes puntos:
- 1.
Los criterios para iniciar el tratamiento en los pacientes con EG1 deben valorarse de forma individualizada y se deben seguir los objetivos terapéuticos a corto y largo plazo recomendados internacionalmente.
- 2.
Miglustat y eliglustat son inhibidores de la GCst, pero presentan distintos mecanismos de acción y propiedades farmacológicas, lo que explica sus diferencias en la efectividad. Esto obliga a utilizarlos según sus indicaciones de ficha técnica y nunca se han de considerar como equivalentes.
- 3.
Se recomienda valorar las indicaciones de cada fármaco antes de iniciar el tratamiento ya que miglustat está indicado para pacientes con EG1 no grave que no pueden recibir otro tratamiento de primera línea, mientras que eliglustat está indicado para pacientes con EG1 de cualquier gravedad, en primera línea y sin necesidad de estabilización previa con TRE.
- 4.
No es necesario administrar TRE previo al inicio de eliglustat, pudiéndose iniciar en pacientes independientemente de la gravedad de la EG1.
- 5.
Eliglustat es un fármaco que se metaboliza a nivel hepático por el citocromo CYP2D6 y, en menor medida, por CYP3A4. Por ello, antes de pautar el tratamiento con eliglustat debemos conocer su fenotipo metabólico respecto a CYP2D6 del paciente. Este paso es crítico en el inicio y dosificación del tratamiento.
- 6.
Se debe evitar el uso de eliglustat durante el embarazo, empleando únicamente el TRE.
- 7.
Antes de iniciar tratamiento con eliglustat, se deberá realizar una evaluación cardiológica y ósea, cuantificando el fenotipo a través de los índices de gravedad y el uso de los biomarcadores. Todos los pacientes deberán someterse a una evaluación regular completa, cuya frecuencia depende de si se han alcanzado los objetivos terapéuticos.
- 8.
No debe ser un objetivo terapéutico la normalización de los biomarcadores.
- 9.
La asociación de eliglustat con fármacos metabolizados a través de los citocromos CYP2D6 y CYP3A4 –o bien que utilicen la Glucoproteína P– se debe realizar teniendo en cuenta al ajuste de dosis en función del fenotipo metabolizador de eliglustat y el grado de inhibición/inducción del fármaco asociado.
- 10.
Eliglustat ha demostrado mejoría en la calidad de vida de los pacientes EG1.
El origen de la iniciativa para la realización del protocolo surgió de un grupo de médicos que tratan a pacientes con EG1. La motivación de la ejecución de estas recomendaciones parte de la necesidad observada de unificar los criterios de actuación y minimizar la variabilidad clínica, para aumentar la seguridad clínica, sin que haya existido otra influencia en el contenido del mismo. A lo largo de la elaboración de este documento, todos los autores han mantenido la independencia editorial y de opinión, habiendo recibido apoyo logístico por la empresa Anima Consulting SL., contratada por Sanofi España, que no mantuvo relación directa con los autores.
Conflicto de interesesLos autores del texto declaran los siguientes conflictos de intereses:
Miguel Ángel Torralba-Cabeza ha recibido fondos para investigación y participado en conferencias financiadas por Genzyme-Sanofi® y Shire®.
Marta Morado-Arias ha recibido honorarios de Genzyme-Sanofi®, Agios®, Alexion®, Novonordisk® y Novartis® por su participación como experto en Advisory Boards y como ponente en conferencias.
Agustín Pijierro-Amador ha recibido honorarios por su participación en conferencias financiadas por Genzyme-Sanofi® y Takeda-Shire®, Alexion® y Alnylan®.
María Cristina Fernández-Canal ha recibido honorarios por su participación en conferencias financiadas por Genzyme-Sanofi®.
Jesús Villarrubia-Espinosa ha recibido fondos para investigación y participado en conferencias financiadas por Genzyme-Sanofi® y Takeda-Shire®.


