


Las porfirias hepáticas agudas (PHA) son un grupo de enfermedades minoritarias que incluyen la porfiria aguda intermitente, la porfiria variegata, la coproporfiria hereditaria y la porfiria por deficiencia de la 5-aminolevulínico ácido deshidratasa. Los síntomas de la PHA son inespecíficos, lo que, junto a su baja prevalencia, hacen que el diagnóstico y el seguimiento de estos pacientes sean complejos.
Materiales y métodosEste proyecto utilizó la metodología DELPHI para responder unas preguntas PICO relacionadas con el manejo de pacientes con PHA. El objetivo fue alcanzar un consenso multidisciplinario entre expertos en porfiria sobre esas preguntas PICO para mejorar el diagnóstico y el seguimiento de los pacientes con PHA.
ResultadosSe definieron diez preguntas PICO y se agruparon en cuatro dominios: 1.Diagnóstico bioquímico de los pacientes con PHA. 2.Estudio molecular de los pacientes con PHA. 3.Seguimiento de los pacientes con PHA. 4.Cribado de complicaciones a largo plazo en los pacientes con PHA.
ConclusionesLas preguntas PICO y la metodología DELPHI han proporcionado un consenso sobre temas relevantes y controvertidos para mejorar el manejo de pacientes con PHA.
Acute hepatic porphyrias (AHPs) are a group of rare diseases that encompasses acute intermittent porphyria, variegate porphyria, hereditary coproporphyria, and 5-aminolaevulinic acid dehydratase deficiency porphyria. Symptoms of AHP are nonspecific which, together with its low prevalence, difficult the diagnosis and follow-up of these patients.
Material and methodsThis project used DELPHI methodology to answer PICO questions related to management of patients with AHPs. The objective was to reach a consensus among multidisciplinary porhyria experts providing answers to those PICO questions for improving diagnosis and follow-up of patients with AHP.
ResultsTen PICO questions were defined and grouped in four domains: 1.Biochemical diagnosis of patients with AHP. 2.Molecular tests for patients with AHP. 3.Follow-up of patients with AHP. 4.Screening for long-term complications of patients with AHP.
ConclusionsPICO questions and DELPHI methodology have provided a consensus on relevant and controversial issues for improving the management of patients with AHP.
El grupo hemo es el componente no aminoacídico de distintas proteínas esenciales para llevar a cabo una amplia gama de funciones vitales, como la hemoglobina, las catalasas o las peroxidasas. El grupo hemo se sintetiza a través de ocho pasos enzimáticos, y las variantes patogénicas o mutaciones en los genes que codifican estas enzimas dan lugar a los ocho subtipos de porfiria1,2. Las porfirias hepáticas agudas (PHA) son un subgrupo de enfermedades minoritarias (ORPHA: 95157) que incluyen la porfiria aguda intermitente (PAI) (la más común), la porfiria variegata (PV), la coproporfiria hereditaria (CPH) y la extremadamente infrecuente porfiria por deficiencia de ácido delta-aminolevulínico (ALA) deshidratasa (ADP)3,4. La PV y la CPH son porfirias mixtas, que presentan además afectación cutánea1-4. En pacientes con PHA, ante la exposición a factores precipitantes, la disminución del grupo hemo por el bloqueo enzimático provoca la inducción de la ALA sintasa1 hepática (ALAS1), lo que, unido al bloqueo enzimático que el paciente presenta por su enfermedad, resulta en la acumulación de ALA y porfobilinógeno (PBG), los llamados precursores de porfirinas. Estos precursores causan daño al sistema nervioso y otros órganos, ocasionando crisis agudas potencialmente mortales y manifestaciones crónicas de la enfermedad2,4.
Aunque sugestivos, los síntomas de la PHA son inespecíficos y tienen una gravedad muy variable. Por ello, la detección de niveles elevados en orina de ALA y PBG son hallazgos clave para el diagnóstico de una crisis aguda2. El test de Hoesch puede detectar la presencia de PBG en la orina de manera sencilla y rápida, convirtiéndose en una prueba cualitativa de primera línea en el cribado de una posible crisis de porfiria. En esta prueba, se agregan dos gotas de orina fresca a 1ml de reactivo de Ehrlich (solución que contiene el indicador de PBG p-dimetilaminobenzaldehído disuelto en ácido clorhídrico). En caso de un aumento de PBG en la muestra de orina, al entrar en contacto con el indicador, se desarrolla inmediatamente un color rojo cereza en todo el tubo después de la agitación5,6. Los precursores de porfirina deben determinarse y cuantificarse específicamente en orina después del test de Hoesch para confirmar el diagnóstico. Para ello, no es necesario una muestra de orina de 24horas, pero debe emplearse una muestra de orina simultánea y deben corregirse las concentraciones de ALA y PBG para evitar sesgos por dilución, mediante su correlación con la concentración de creatinina en la muestra obtenida5,7.
Los síntomas y el curso clínico de la PHA son muy variables, dificultando el diagnóstico y el seguimiento de estos pacientes. De hecho, Waldenström describió la porfiria aguda como la «pequeña imitadora» en 1937, contrastándola con la sífilis, la «gran imitadora» de principios del sigloxx8. En este sentido, nuestro estudio tiene como objetivo buscar un consenso entre expertos en porfiria para mejorar el diagnóstico y el seguimiento de los pacientes con PHA.
Materiales y métodosDiseño del estudioSe trata de un trabajo multicéntrico y multidisciplinario entre expertos en el manejo de pacientes con PHA. La duración del proyecto fue de 6meses, desde junio hasta diciembre de 2023. El estudio fue aprobado por el Comité de Ética de Investigación Clínica del Hospital Universitari de Bellvitge (Barcelona, España; número de aprobación PR280/23).
El objetivo principal fue alcanzar un consenso sobre la prevención, el diagnóstico y el seguimiento de pacientes con PHA en diferentes escenarios clínicos controvertidos, excluyendo el tratamiento farmacológico.
La metodología utilizada estableció un conjunto de problemas clínicos mediante preguntas PICO (del inglés Patient, Intervention, Comparison, Outcomes) y después se aplicó el método DELPHI para elaborar las respuestas9-11. DELPHI es una metodología estructurada que recopila sistemáticamente opiniones de expertos sobre un problema específico y permite construir un consenso del grupo10,11. La característica clave que permite llegar al consenso es que los participantes reciben retroalimentación sobre sus respuestas y pueden irlas ajustando y acordando mediante un proceso iterativo10,11. Los beneficios de la combinación de preguntas PICO y la metodología DELPHI se han descrito ampliamente en estudios previos9-12.
Un Comité Científico (AR-M, EG-N y MM-C) reclutó un panel de seis expertos (JSGM, JCF, MEH-C, PAP, JJ y FMV) en el manejo de pacientes con porfiria de diferentes centros de España. El proyecto se desarrolló en cuatro fases. En la fase1 se definieron los dominios y las preguntas PICO. En la fase2 se discutieron y respondieron a las preguntas PICO en tres rondas grabadas. En la fase3, mediante varias rondas de retroalimentación con todos los expertos, se redactó una respuesta global para cada pregunta PICO. El panel de expertos utilizó los términos se «recomienda» o se «sugiere» en función de que la fuerza de la recomendación fuera fuerte o débil, de acuerdo al grado en el que se puede tener confianza en que los efectos deseables de una intervención superan a sus efectos indeseables13. Finalmente, en la fase4, cada miembro del panel valoró individualmente si estaba de acuerdo, en desacuerdo o ni de acuerdo ni en desacuerdo con las respuestas a las preguntas PICO, y se recopiló el grado de consenso según las frecuencias absolutas de dicha valoración.
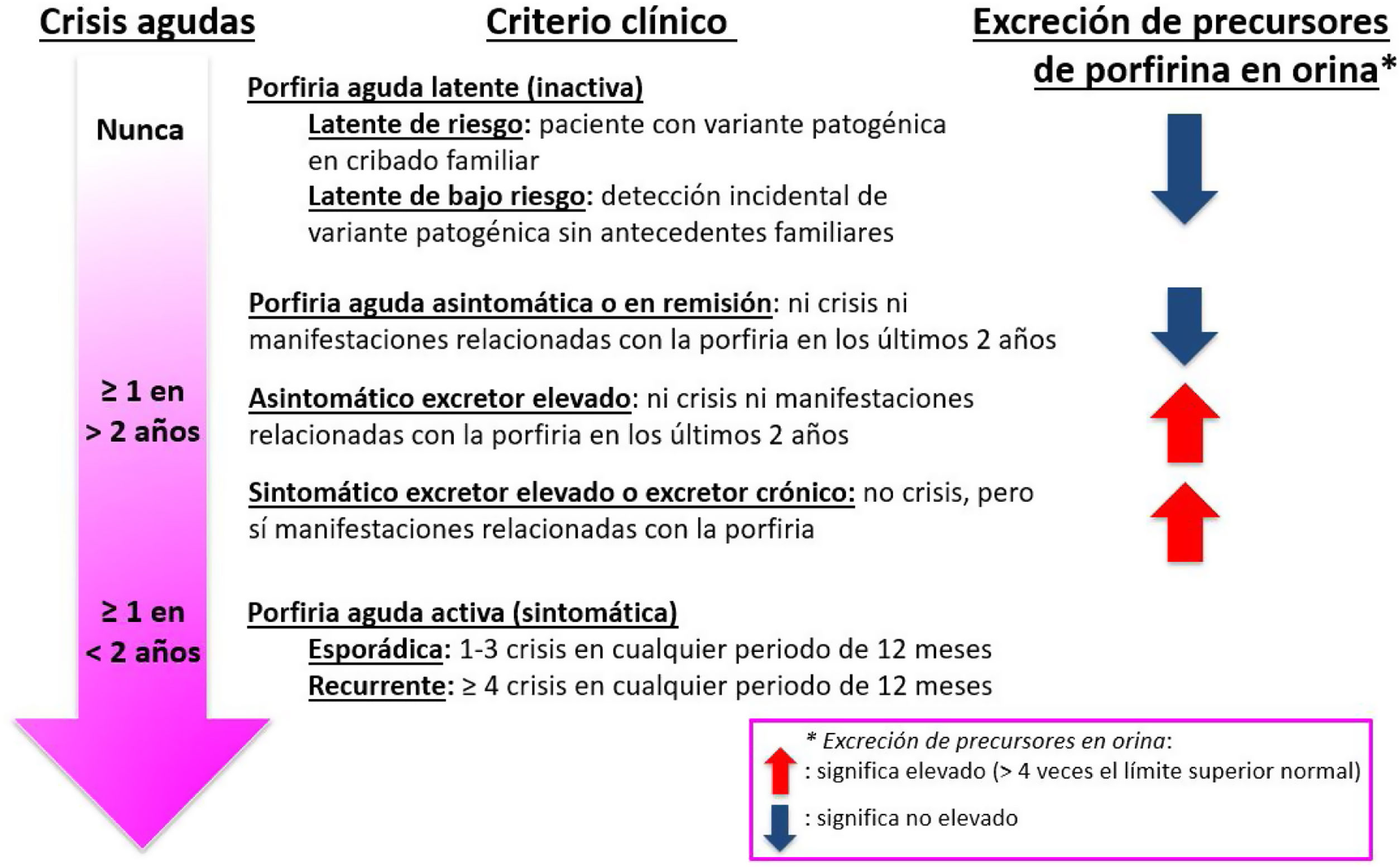
Terminología utilizadaEl panel utilizó algunos términos clave y definiciones recientemente establecidos por la Red Europea de Porfiria (European Porphyria Network [EPN]) para los términos relacionados con el diagnóstico, el tratamiento y el seguimiento de los pacientes con PHA7. Estas definiciones de los patrones típicos de porfiria según los síntomas y la excreción de precursores en orina se muestran en la figura 1. Así, se adoptó la definición de crisis aguda de porfiria como aquel episodio que incluye ≥2 de las siguientes manifestaciones: dolor intenso (principalmente en el abdomen), náuseas, vómitos, estreñimiento, retención o incontinencia urinaria, hipertensión arterial sistémica, taquicardia, hiponatremia, neuropatía periférica o afectación del sistema nervioso central (por ejemplo, convulsiones, psicosis o síndrome de encefalopatía posterior reversible), típicamente persistiendo durante más de 24horas en ausencia de otras explicaciones probables, junto con elevación de precursores (ALA y PBG) en orina. Se definió una crisis aguda de porfiria como grave si estaba asociada con ≥1 de las siguientes características: retención o incontinencia urinaria, hiponatremia significativa, arritmias, neuropatía periférica o afectación del sistema nervioso central. El panel de expertos se refirió como fenotipo bioquímico compatible aquel que incluye la excreción aumentada de precursores de porfirinas (ALA y PBG) en orina y de porfirinas tanto en orina como en heces (en este último caso, en las porfirias mixtas, PV y CPH). Este fenotipo bioquímico completo permite realizar el diagnóstico bioquímico entre los diferentes subtipos de porfiria.
Resultados
Se definieron 10 preguntas PICO divididas en cuatro dominios (tabla 1).
Dominios con sus respectivas preguntas PICO
| Dominio | Preguntas PICO |
|---|---|
| Dominio 1. Diagnóstico bioquímico de los pacientes con PHA | PICO 1. ¿En qué pacientes atendidos en servicios de urgencias por dolor abdominal deberíamos realizar el test de Hoesch?P: Pacientes adultos jóvenes (15-50 años) atendidos en servicios de urgencias por dolor abdominal.I: Realizar el test de Hoesch.C: No realizarlo.O: Diagnóstico de PHA. |
| PICO 2. En el diagnóstico diferencial de la neuropatía axonal sensitivo-motora aguda/subaguda progresiva ¿debemos incluir la PHA y qué test inicial debemos realizar?P: Pacientes con neuropatía atípica tipo síndrome de Guillain-Barré.I: Realizar el test de Hoesch y determinación de precursores de porfirinas en orina.C: No realizarlo.O: Diagnóstico de PHA. | |
| PICO 3. ¿Cuál debería ser el siguiente paso diagnóstico en aquellos pacientes con sospecha de PHA y un test de Hoesch negativo?P: Paciente con sospecha clínica de PHA y un test de Hoesch negativo.I: Realizar un estudio bioquímico cuantitativo de precursores en orina.C: No realizarlo.O: Diagnóstico de PHA. | |
| Dominio 2. Estudio molecular de los pacientes con PHA | PICO 4. ¿Qué estudio complementario se debería realizar en pacientes con fenotipo clínico y bioquímico compatible con PHA?P: Paciente con sospecha clínica de PHA y perfil bioquímico compatible.I: Realizar estudio genético de los genes asociados (HMBS, CPOX, PPOX y ALAD) mediante secuenciación masiva (panel/exoma clínico) o Sanger.C: No realizarlo.O: Confirmación de PHA. |
| PICO 5. ¿Qué estudios deberíamos realizar en pacientes con perfil clínico-bioquímico compatible con PHA y secuenciación masiva de los genes asociados negativa o no concluyente?P: Paciente con síntomas y alteración de parámetros bioquímicos compatibles con PHA y estudio genético negativo o no concluyente.I: Realizar otros estudios genéticos según patrón bioquímico, en un centro de referencia en genética.C: No realizarlo.O: Diagnóstico genético de PHA. | |
| PICO 6. ¿Qué tipo de estudio complementario de primera línea hay que realizar en los familiares de un paciente con PHA caracterizado genéticamente?P: Familiares asintomáticos de un paciente índice diagnosticado de PHA.I: Realizar estudio genético de la variante familiar asociada a PHA.C: No realizarlo.O: Diagnóstico de PHA en pacientes asintomáticos. | |
| Dominio 3. Seguimiento de los pacientes con PHA | PICO 7. ¿Se debe realizar un seguimiento que incluya determinación de precursores de porfirinas en orina de los pacientes con PHA?P: Pacientes con PHA sintomáticos y asintomáticos.I: Realizar seguimiento bioquímico de precursores de porfirinas en orina periódicamente.C: Realizar exclusivamente seguimiento clínico.O: Valorar el grado de actividad de la PHA. |
| PICO 8. En un paciente con PHA que consulta por dolor abdominal, ¿la positividad en el test de Hoesch es siempre diagnóstico de una crisis aguda?P: Pacientes con diagnóstico de PHA y sospecha de crisis aguda.I: Basarse exclusivamente en el test de Hoesch.C: Comparar los niveles de precursores de porfirina (ALA/PBG) en orina de ese momento con su basal y constatar un aumento significativo.O: Diagnóstico / exclusión ante la sospecha de crisis aguda. | |
| Dominio 4. Cribado de complicaciones a largo plazo en los pacientes con PHA | PICO 9. ¿Debe realizarse un cribado del hepatocarcinoma (HCC) en los pacientes con PHA?P: Pacientes con PHA, sintomáticos y asintomáticos.I: Realizar ecografía periódica (cada 6-12 meses).C: No realizarla.O: Diagnóstico precoz de HCC. |
| PICO 10. ¿Debe realizarse monitorización periódica de la presión arterial (PA) y la función renal en los pacientes con PHA?P: Pacientes con PHA sintomáticos y asintomáticos.I: Realizar seguimiento periódico (anual) de cifras de PA y función renal (creatinina y tasa de filtración glomerular).C: No realizar dichas determinaciones periódicas.O: Diagnóstico y tratamiento precoz de hipertensión arterial o insuficiencia renal. |
PHA: porfiria hepática aguda; PICO: Patient, Intervention, Comparison, Outcomes.
PICO 1. ¿En qué pacientes atendidos en servicios de urgencias por dolor abdominal deberíamos realizar el test de Hoesch?
- 1.
En aquellos pacientes con antecedentes de dolor abdominal recurrente o incluso si es el primer episodio con dolor intenso o grave (por ejemplo, con hiponatremia o afectación neurológica), se recomienda realizar el test de Hoesch como parte del estudio etiológico inicial.
- 2.
Se sugiere realizar el test de Hoesch en adultos jóvenes (15-50años) atendidos en urgencias por dolor abdominal, sin un diagnóstico específico después de haberse realizado un estudio etiológico razonable.
Consenso: de acuerdo 9/9.
Comentarios. La simplicidad del test de Hoesch lo convierte en una herramienta valiosa en entornos de urgencias, donde un diagnóstico rápido es esencial para iniciar un tratamiento adecuado5,6. El dolor abdominal es el síntoma más común de una crisis aguda y suele ser grave, generalizado y acompañado de náuseas, vómitos, estreñimiento o diarrea14. La PHA debe considerarse en el diagnóstico diferencial del dolor abdominal, especialmente cuando una evaluación diagnóstica inicial no sugiere una causa más común, incluso en pacientes varones. Después de un resultado positivo en el test de Hoesch, es imprescindible la determinación cuantitativa del fenotipo bioquímico compatible para confirmar el diagnóstico e identificar el subtipo de porfiria5-7.
PICO 2. En el diagnóstico diferencial de la neuropatía axonal sensitivo-motora aguda/subaguda progresiva ¿debemos incluir la PHA y qué test inicial debemos realizar?
- 1.
En pacientes con neuropatía axonal sensitivo-motora aguda/subaguda progresiva tipo Guillain-Barré con características atípicas se recomienda realizar el test de Hoesch para acelerar el diagnóstico y poder administrar el tratamiento adecuado.
- 2.
Si hay una alta sospecha clínica, se recomienda solicitar simultáneamente un estudio cuantitativo de precursores (ALA y PBG) en orina, incluso si el resultado del test de Hoesch resultara negativo.
Consenso: de acuerdo 9/9.
Comentarios. La neuropatía periférica en la PHA puede ser motora, sensitiva o mixta, y los síntomas son altamente variables, incluyendo dolor en las extremidades, debilidad muscular o pérdida de sensibilidad. Aunque poco común, la PHA también puede manifestarse como una parálisis ascendente progresiva aguda, simulando el síndrome de Guillain-Barré15,16. El inicio puede ser simétrico o asimétrico y afectar proximalmente los brazos o las piernas. Esta condición puede progresar a cuadriplejia e insuficiencia respiratoria, requiriendo ingreso en una unidad de cuidados intensivos15,16.
Es importante destacar que la PHA es tratable y, por tanto, la potencial discapacidad se puede prevenir si la neuropatía se diagnostica y trata precozmente. Puede haber señales de sospecha de PHA como causa de los síntomas neurológicos, como antecedentes recientes de dolor abdominal, hiponatremia o factores precipitantes conocidos2,17. El test de Hoesch puede ser una herramienta inicial y eficiente en el proceso diagnóstico5,6.
PICO 3. ¿Cuál debería ser el siguiente paso diagnóstico en aquellos pacientes con sospecha de PHA y un test de Hoesch negativo?
- 1.
Si persiste una alta sospecha clínica, se recomienda cuantificar la excreción urinaria de precursores de porfirinas (ALA y PBG) antes de descartar una crisis aguda de porfiria.
Consenso: de acuerdo 9/9.
Comentarios. En pacientes con una alta sospecha clínica se necesitan pruebas adicionales para confirmar el diagnóstico y el tipo de porfiria, incluso si el resultado del test de Hoesch es negativo5-7. De hecho, pequeños aumentos en PBG pueden pasar desapercibidos al utilizar esta prueba. A pesar de ello, esta es una situación muy inusual en pacientes con una crisis aguda, ya que presentan niveles marcadamente elevados de PBG en orina (en el rango de 10-20 veces más altos)5,6. Otros posibles falsos negativos del test de Hoesch incluyen problemas técnicos, aunque son muy poco comunes en esta prueba tan sencilla5. Hay que considerar también que el test de Hoesch es negativo en la ADP y además en la tirosinemia hereditaria tipoI y en algunas entidades con síntomas superponibles a la PHA, como el saturnismo, donde solo se produce elevación de ALA5,14,18.
A pesar de estas situaciones poco comunes, la fiabilidad de la prueba depende del escenario clínico. Si hay dolor abdominal grave, un resultado negativo debería orientar a otras causas distintas de la PHA. Sin embargo, en mujeres jóvenes, un resultado negativo debería confirmarse con la cuantificación de la excreción urinaria de precursores antes de descartar la porfiria2,4,5,7.
Dominio 2. Estudio molecular de los pacientes con PHAPICO 4. ¿Qué estudio complementario se debería realizar en pacientes con fenotipo clínico y bioquímico compatible con PHA?
- 1.
En estos pacientes se recomienda realizar estudio molecular mediante secuenciación masiva (panel específico o exoma clínico).
- 2.
Si el paciente procede de una zona en la que existe una variante genética patogénica con efecto fundador asociada a PHA, el estudio directo de dicha variante mediante secuenciación Sanger puede resultar coste-efectivo.
Consenso: de acuerdo 9/9.
Comentarios. En pacientes con fenotipo bioquímico compatible con PHA, la confirmación diagnóstica se establece mediante estudio genético. La realización de dicho estudio sin conocer el perfil clínico-bioquímico puede conllevar problemas de interpretación de las variantes genéticas identificadas4. En pacientes sin antecedentes familiares estudiados se recomienda la realización de secuenciación masiva (next generation sequencing [NGS]) que incluya al menos los cuatro genes asociados a la PHA (HMBS, CPOX, PPOX y ALAD), mediante un panel específico o exoma clínico. Con este estudio se caracterizan molecularmente entre el 95 y el 99% de los casos de porfiria18,19. El gen implicado con mayor frecuencia es el gen HMBS, relacionado con la PAI. La penetrancia actual de este trastorno se desconoce, pero se estima en torno al 1% en portadores asintomáticos, y superior al 20% en aquellas familias con pacientes que manifiestan la enfermedad20,21. En el estudio inicial de pacientes procedentes de áreas con efecto fundador identificado, el estudio directo de la variante patogénica mediante secuenciación Sanger resulta actualmente coste-efectivo19-22. Asimismo, está indicado realizar asesoramiento genético antes y después de realizar el test genético.
PICO 5. ¿Qué estudios deberíamos realizar en pacientes con perfil clínico-bioquímico compatible con PHA y secuenciación masiva de los genes asociados negativa o no concluyente?
- 1.
Si el perfil clínico y bioquímico es compatible con PHA y no se ha identificado su base genética por secuenciación masiva, se recomienda derivar a un centro de referencia en genética con personal experto en PHA para su estudio avanzado.
Consenso: de acuerdo 9/9.
Comentarios. El supuesto que exista una evidencia clínica y bioquímica de PHA y que la secuenciación sea negativa o no concluyente, es muy poco frecuente (<5%)19. Si la sospecha clínica-bioquímica persiste, se deberán realizar otros estudios genéticos, como la amplificación de sondas múltiples dependientes de ligación (Multiplex Ligation dependent Probe Amplification [MLPA]) para descartar deleciones/duplicaciones, secuenciación de lectura larga para cribado de alteraciones estructurales, matriz de hibridación genómica comparada (comparative genomic hybridisation array [aCGH]), pruebas funcionales para variantes de significado clínico incierto o estudios enzimáticos específicos, que permitan identificar su base molecular23. Para ello es necesaria la derivación de las muestras a un centro de referencia en genética con personal experto en PHA, para la caracterización completa de estos pacientes.
PICO 6. ¿Qué tipo de estudio complementario de primera línea hay que realizar en los familiares de un paciente con PHA caracterizado genéticamente?
- 1.
Se recomienda ofrecer estudio genético a los familiares (tanto sintomáticos como asintomáticos) de un paciente con PHA de la variante genética causal identificada, y en caso de resultado positivo hacer el estudio bioquímico basal.
Consenso: de acuerdo 9/9.
Comentarios. En el estudio de segregación familiar con variante genética patogénica conocida, el estudio directo mediante secuenciación Sanger resulta actualmente coste-efectivo22. En los portadores identificados mediante el estudio genético familiar se recomienda una determinación bioquímica cuantitativa basal de precursores de porfirinas (ALA y PBG) en orina, ya que existen portadores latentes excretores. También está indicado realizar asesoramiento genético antes y después de realizar el test genético.
Aunque el paciente pueda no desarrollar nunca la enfermedad, dada la importancia de los factores ambientales desencadenantes en el desarrollo de las crisis, deberían aportarse recomendaciones sobre fármacos y otras situaciones que las pueden precipitar22,24. Tener conciencia de ser portador de PHA disminuye la frecuencia de aparición de crisis25.
Dominio 3. Seguimiento de los pacientes con PHAPICO 7. ¿Se debe realizar un seguimiento que incluya determinación de precursores de porfirinas en orina de los pacientes con PHA?
- 1.
Se recomienda realizar un seguimiento anual con determinación de precursores de porfirinas en orina de todos los pacientes que han sufrido al menos una crisis a lo largo de su vida y de los pacientes con excreción urinaria aumentada de precursores.
- 2.
Se sugiere realizar un seguimiento bianual a los pacientes con porfiria latente o inactiva con determinación de precursores de porfirinas en orina.
Consenso: de acuerdo 9/9.
Comentarios. Los niveles de ALA y PBG se relacionan con la actividad de la enfermedad y el desarrollo de complicaciones a largo plazo26. Por ello, es necesario realizar un seguimiento de los pacientes con PHA, que se debe adecuar en función de la clínica y la expresión bioquímica7. Aunque no se ha especificado la forma y el tiempo de cómo realizarlo, se propone la siguiente estrategia de seguimiento:
- -
Tras un ingreso por una crisis porfírica sería adecuado que la revisión en la consulta sea en el mes siguiente al alta para reevaluar los factores precipitantes, las medidas preventivas y el adecuado control de los síntomas.
- -
Los pacientes que han tenido al menos una crisis aguda en su vida, o aquellos que presentan una excreción aumentada de precursores de porfirina en orina, tienen mayor probabilidad de sufrir una crisis o complicaciones a largo plazo4. Por ese motivo, se benefician de un seguimiento adecuado en una unidad de referencia con determinación anual de precursores4,7. Este seguimiento podrá ser más frecuente según las necesidades clínicas de cada paciente27.
- -
Los pacientes que nunca han presentado una crisis (porfiria latente o inactiva) no necesitan un seguimiento clínico estrecho. A pesar de ello, también se les debe aportar educación en salud, informando de los signos de alarma y de los factores precipitantes de la PHA27.
PICO 8. En un paciente con PHA que consulta por dolor abdominal, ¿la positividad en el test de Hoesch es siempre diagnóstico de una crisis aguda?
- 1.
En los pacientes con PHA que acuden a urgencias por un episodio de dolor abdominal se recomienda hacer una valoración completa para descartar diagnósticos alternativos.
- 2.
Para una valoración óptima del resultado del test de Hoesch, se recomienda la determinación cuantitativa de precursores de porfirinas en orina y su comparación con la basal realizada durante el seguimiento.
Consenso: de acuerdo 9/9.
Comentarios. En pacientes con diagnóstico previo de PHA que consultan a urgencias por dolor abdominal es fundamental estar atentos a la presencia de síntomas que nos apoyen el diagnóstico, pero también descartar diagnósticos alternativos4,28. En estos pacientes, la normalidad de los niveles de ALA y PBG descarta la PHA como etiología de los síntomas29.
En el caso de que estemos ante un paciente con antecedente de PHA sintomática o asintomática pero no excretores, la positividad de un test de Hoesch puede ser la primera prueba de cribado sugestiva de nueva crisis aguda4-6. Sin embargo, esta prueba carece de utilidad en los excretores crónicos, dado que en ellos el test de Hoesch es persistentemente positivo7. Si se desconoce si un paciente con PHA es excretor elevado o no, hay que recordar que es poco frecuente que los pacientes con CPH o PV mantengan una excreción elevada de precursores, por lo que en estos pacientes la positividad en el test de Hoesch puede ser sugerente de una crisis porfírica30.
Por otro lado, aunque el aumento significativo de precursores sobre su nivel basal apoya el diagnóstico de crisis aguda, esta determinación no suele estar disponible en el momento agudo7. Por ello, en un paciente con PHA que presenta un cuadro sugerente de una crisis aguda con test de Hoesch positivo y ante la ausencia de un diagnóstico alternativo, debería asumirse el diagnóstico de una crisis porfírica e iniciar tratamiento sin esperar el resultado de la cuantificación de precursores en orina.
Dominio 4. Cribado de complicaciones a largo plazo en los pacientes con PHAPICO 9. ¿Debe realizarse un cribado del hepatocarcinoma (HCC) en los pacientes con PHA?
- 1.
Se recomienda hacer una ecografía abdominal cada 6 o 12meses como cribado del HCC en los pacientes mayores de 50años con porfiria activa sintomática.
- 2.
En los pacientes asintomáticos no excretores o con porfiria latente, se sugiere prescindir de dicha ecografía.
Consenso: de acuerdo 9/9.
Comentarios. Los pacientes con PHA presentan un mayor riesgo de HCC, con una prevalencia entre el 1,5 y el 1,8%, siendo más frecuente en los mayores de 50años4,31-33. Por todo ello, se recomienda la realización de una ecografía cada 6 o 12meses, pudiéndose individualizar según la edad, la presencia de crisis o la excreción crónica de precursores. Así, se recomienda hacer una ecografía abdominal cada 6meses en los pacientes mayores de 50años. A pesar de la falta de evidencia, se podría realizar una monitorización menos estrecha (cada 12meses), intercalando la determinación de alfa-fetoproteína cada 6meses34. En los pacientes jóvenes asintomáticos no excretores o con porfiria latente se podría prescindir de la ecografía para el cribado de HCC, salvo que cambiase la situación clínica, siempre individualizando según la edad y los factores de riesgo (obesidad, esteatosis, infección viral…). En la evaluación de este riesgo se podría considerar también la determinación anual de enzimas hepáticas, ya que se ha reportado su elevación hasta en un 28% de estos pacientes y podría identificar una población de riesgo para el desarrollo de complicaciones a largo plazo, como el HCC4,35.
PICO 10: ¿Debe realizarse monitorización periódica de la presión arterial (PA) y la función renal en los pacientes con PHA?
- 1.
Se recomienda realizar una monitorización de la PA y de la función renal al menos una vez al año en todos los pacientes con porfiria activa (esporádica o recurrente).
- 2.
Se sugiere realizar una monitorización de la PA y de la función renal cada 1-2años en los pacientes con excreción elevada de precursores, sintomáticos o asintomáticos, sobre todo si se acompaña de otros factores de riesgo cardiovascular, o en mayores de 50años.
Consenso: de acuerdo 9/9.
Comentarios. Los pacientes con PHA presentan mayor riesgo de desarrollar hipertensión arterial o insuficiencia renal, con una incidencia superior al 40% y al 20%, respectivamente33. En el estudio internacional y prospectivo EXPLORE, que evaluó la historia natural de 112 pacientes con PHA, el 68% presentaron deterioro del filtrado glomerular (<90ml/min/1,73m2)36.
El desarrollo de hipertensión arterial e insuficiencia renal en la PHA se considera multicausal. Por un lado, los precursores de porfirinas ejercen un daño directo sobre las células renales de los túbulos proximales. Por otro, en las crisis agudas los precursores también generan vasoconstricción, que, junto con la activación del sistema simpático, produce un ascenso de la PA. También existe evidencia que la variante genética del transportador-2 de péptidos (PEPT-2) confiere una mayor afinidad por el ALA a nivel del túbulo proximal, causando reabsorción del mismo y contribuyendo al daño renal crónico37.
La determinación de la función renal (filtrado glomerular y/o cociente de albúmina/creatinina) y la PA son sumamente sencillas, por lo que deben de ser monitorizadas de forma regular. Esta monitorización será más o menos estrecha en función del número de crisis, de la excreción urinaria de precursores o de la presencia de otros factores de riesgo cardiovascular33.
Limitaciones y conclusiónEste estudio presenta algunas limitaciones. En primer lugar, la selección de expertos fue arbitraria. Sin embargo, se incluyeron diferentes especialistas con amplia experiencia en el campo de la porfiria en diversos entornos. En segundo lugar, las limitaciones de la metodología DELPHI ya han sido descritas anteriormente9-12. A pesar de esto, los criterios de consenso fueron definidos previamente y la amplia mayoría de consenso obtenido en las preguntas PICO respalda la solidez de las recomendaciones. Finalmente, la evidencia científica en este campo está evolucionando, por lo que las recomendaciones actuales pueden modificarse según los resultados de nuevos ensayos clínicos aleatorizados.
En conclusión, la combinación de preguntas PICO y la metodología DELPHI proporciona un consenso sobre temas relevantes y controvertidos acerca del abordaje de los pacientes con porfiria, que permite mejorar su diagnóstico y seguimiento.
FinanciaciónEste estudio ha recibido apoyo técnico institucional y ayuda económica no condicionada de Alnylam, que no tuvo implicación alguna en su conceptualización o diseño ni en el contenido del manuscrito para su publicación.
Consideraciones éticasEste estudio ha sido aprobado por el Comité de Ética de Investigación Clínica del Hospital Universitari de Bellvitge (Barcelona, España) con el número de aprobación PR280/23.
Conflicto de interesesAntoni Riera-Mestre ha recibido financiación por presentaciones, proyectos de investigación y apoyo para congresos de Sanofi, Takeda, Rovi, Bayer Healthcare, Alnylam, BMS-Pfizer y Daichii Sanchyo.
José Salvador García Morillo ha recibido financiación por presentaciones, proyectos de investigación y apoyo para congresos de Sanofi, Alnylam, MSD, Roche y GSK.
Javier Castelbón Fernández ha recibido financiación por presentaciones y apoyo para congresos de Alnylam.
María Encarna Hernández-Contreras ha recibido financiación por presentaciones y apoyo para congresos de Alnylam y Takeda.
Paula Aguilera Peiró no declara conflicto de intereses.
Javier Jacob ha recibido financiación por presentaciones, proyectos de investigación y apoyo para congresos de Sanofi, Takeda, Rovi, Bayer Healthcare, Alnylam, BMS-Pfizer y Daichii Sanchyo.
Fernando Martínez Valle no declara conflicto de intereses.
E. Guillén-Navarro ha recibido financiación por presentaciones, asesoramiento, proyectos de investigación y apoyo para congresos de Alnylam, Takeda, Biomarin y UCB.
Montse Morales-Conejo ha recibido financiación por presentaciones, proyectos de investigación y apoyo para congresos de Alnylam, Takeda, Sanofi y Chiesi.
Con el apoyo institucional de CERCA Programme / Generalitat de Catalunya.


