


1928 - ESTUDIO DESCRIPTIVO DE UNA FAMILIA AFECTA DE ENFERMEDAD DE VON HIPPEL LINDAU (VHL) EN UN HOSPITAL DE TERCER NIVEL
Hospital Universitario Son Espases, Palma de Mallorca.
Objetivos: Describir las características clínicas de los pacientes con Von Hippel Lindau.
Métodos: Estudio descriptivo, observacional, retrospectivo de una familia con Síndrome de Von Hippel Lindau (VHL) controlada en consultas de Enfermedades Minoritarias del Servicio de Medicina Interna del Hospital Universitario Son Espases.
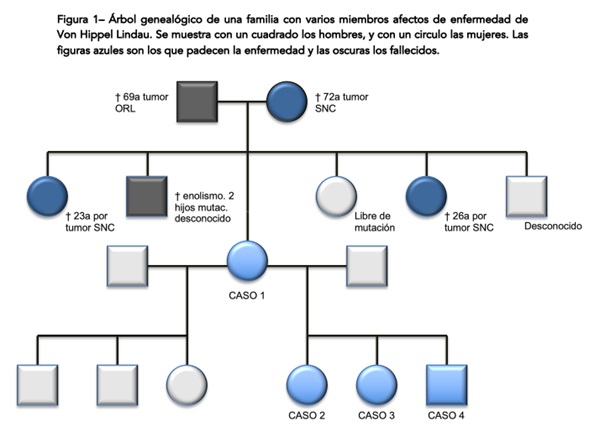
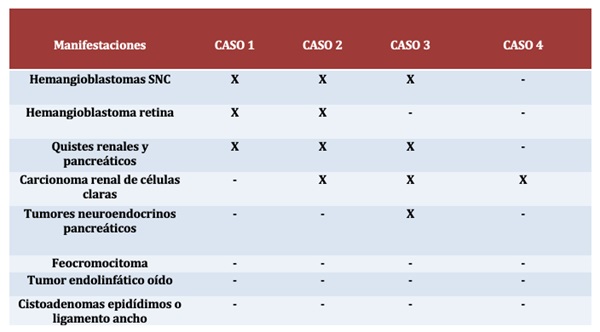
Resultados: Caso 1: mujer de 69 años diagnosticada de VHL, presenta como antecedentes familiares madre fallecida a los 72 años por un tumor del sistema nervioso central (SNC) no especificado. A los 31 años inicia ataxia y cefalea, diagnosticándose hemagioblastoma cerebeloso que pudo ser intervenido. Se realizó estudio genético encontrándose gen VHL mutado, c463+1G>7 en exón 2 del cromosoma 3 en heterocigosis, confirmándose el diagnóstico. Durante el seguimiento ha presentado: angiomas retinianos, hemangioblastomas cerebelosos y medulares, múltiples tumores serosos quísticos pancreáticos y quistes renales bilaterales. La paciente tiene 5 hermanos, 3 de ellos fallecidos, dos hermanas por tumores en SNC a los 26 y 23 años, una hermana libre de mutación y otro hermano que rechazó estudio genético pero no ha presentado clínica. Tiene 6 hijos, 3 con una pareja libres de mutación, y 3 con otra pareja que sí presentan la mutación. Caso 2: mujer de 46 años diagnosticada en el 2013 a raíz de estudio genético, presentando la mutación familiar. Hemangioblastoma cerebeloso en 1997 intervenido. En 2016 se detectó carcinoma renal múltiple de células claras (CRCC) en riñón izquierdo, tratado con nefrectomía parcial. Durante seguimiento nuevo CRCC en 2020 en riñón derecho con nueva nefrectomía parcial. Presenta hemangioblastomas medulares a nivel cervical y dorsal. Como otras manifestaciones: angiomas retinianos, quistes pancreáticos y renales bilaterales. Exámenes ORL normales y metanefrinas anuales negativas hasta el momento. Tiene dos hijos, una hija libre de mutación y un hijo portador. Caso 3: mujer de 40 años diagnosticada también en 2013 a raíz de estudio genético. Afecta de CRCC en 2013 tratado con tumorectomía renal izquierda. Nuevo CRCC múltiple en 2018 que se resecó con doble tumorectomía renal derecha sin signos de recidiva actualmente. Hemangioblastoma bulbar en 2017 intervenido. Tumor neuroendocrino de cabeza de páncreas intervenido en 03/2022 con duodenopancreatectomía cefálica sin precisar radioterapia ni quimioterapia. Como otras manifestaciones: hemangiomas hepáticos y quistes pancreáticos. Tiene 2 hijos con estudio genético negativo. Caso 4: varón de 46 años, portador de la mutación familiar. En 2013 nefrectomía bilateral por CRCC. En 2014 laminectomía por hemangioblastoma medular. Presenta ERC estadio 5 secundaria a binefrectomía, se realiza trasplante renal en 2016. En 08/2021 se realiza esplenopancreatectomía coprocaudal por recidiva pancreática de CRCC. En TC de control en 02/2022 se evidencia progresión de enfermedad, presentando afectación pulmonar, en lecho quirúrgico, estómago y hepática, iniciándose tratamiento con sunitinib.
Discusión: La enfermedad de von Hippel-Lindau (VHL) es un síndrome hereditario autosómico dominante causado por mutaciones en el gen supresor tumoral VHL localizado en el cromosoma 3p25-26, con elevada penetrancia (> 95%) y gran variabilidad fenotípica incluso dentro de la misma familia. La enfermedad consiste en la aparición de múltiples tumores en diferentes órganos a lo largo de toda la vida del individuo. En cuanto a los tumores que pueden aparecer, pueden ser tanto benignos como malignos: hemangioblastomas retinianos, cerebelosos y medulares, cistoadenomas pancreáticos, tumores del saco endolinfático y cistoadenomas del epidídimo y ligamento ancho, tumores neuroendocrinos pancreáticos, feocromocitomas, quistes renales y carcinomas renales de células claras (CRCC). Las principales causas de muerte de estos pacientes son el CRCC metastásico y el hemangioblastoma del SNC. El diagnóstico implica también a los familiares que deben ser estudiados presenten o no sintomatología. Todavía no se ha conseguido establecer una correlación genotipo-fenotipo, los portadores deben continuar seguimiento multidisciplinar y periódico.
Conclusiones: La enfermedad VHL es una enfermedad minoritaria, de baja prevalencia (1:36.000), habitualmente infradiagnosticada y con alto porcentaje de pacientes sin seguimiento. Debe enfatizarse la necesidad de conocer sus manifestaciones, ya que su diagnóstico precoz y seguimiento posterior, mejora notablemente la mediana de supervivencia. Destacar la importancia de realizar un abordaje multidisciplinar y la necesidad de cribaje clínico y genético de los familiares.


