


193 - DESCRIPCIÓN DE UNA POBLACIÓN DE PACIENTES CON ESCLEROSIS TUBEROSA EN UN HOSPITAL TERCIARIO
1Hospital General Universitario Gregorio Marañón, Madrid. 2Universidad Complutense de Madrid, Madrid.
Objetivos: Estudio descriptivo, observacional y retrospectivo de los hallazgos clínicos, radiológicos y genéticos de los pacientes mayores de 18 años con diagnóstico definitivo de esclerosis tuberosa (ET) en seguimiento en una consulta de Enfermedades Minoritarias de Medicina Interna en un hospital de tercer nivel.
Métodos: Revisión de historias clínicas consecutivas de pacientes adultos con diagnóstico de esclerosis tuberosa entre enero de 2012 y enero de 2022 y análisis de los datos recogidos mediante los programas SPSS® 22 (IBM) y Epidat® 4.2 (SERGAS).
Resultados: De los 21 pacientes seleccionados, 14 fueron mujeres (66,7%) y 7 varones (33,3%), con una edad media de 39,3 años (rango 20-69). La media de edad al diagnóstico de ET fue de 13,5 años (rango 0-49). Las primeras manifestaciones de la enfermedad, por orden de frecuencia, fueron las crisis epilépticas (38,1%), las lesiones cutáneas (33,3%) y los rabdomiomas cardíacos (9,5%). En cuanto a la herencia, únicamente un 14,3% de los pacientes tenía antecedentes familiares de ET. Se obtuvo estudio genético completo en el momento del estudio en el 42,8% de los casos, siendo positivo en un 56% (en todos ellos el gen afecto fue TSC2). Respecto a las manifestaciones clínicas a lo largo del curso de la ET, el 90,5% de los pacientes estudiados presentaron manifestaciones dermatológicas: angiofibromas faciales (81%), máculas hipomelanóticas (57,1%), fibromas ungueales (38,1%), placas de Shagreen (19%) y placas fibrosas cefálicas (14,3%). El 90,5% de los pacientes presentaba afectación del sistema nervioso central con túberes corticales como hallazgo más prevalente (90,5%), seguidos de nódulos subependimarios (SEN) (85,7%), líneas de migración radial de la sustancia blanca (76,2%) y astrocitomas subependimarios de células gigantes (14,3%). El 47,6% presentó epilepsia, de los cuales, en el 80% las crisis fueron la manifestación de debut de la enfermedad. Otro 47,6% presentó trastornos neuropsiquiátricos asociados a ET. Los hamartomas retinianos se detectaron en el 19% de los pacientes, uno de los cuales desarrolló glaucoma. Cinco pacientes presentaron rabdomiomas cardíacos (23,8%), en ningún caso con complicaciones. A nivel pulmonar, se detectó linfangioleiomiomatosis (LAM) en el 42,9% de los pacientes, todos ellos de mujeres, con una frecuencia de 85,7% en ≥ 40 años. Por otro lado, se detectó hiperplasia micronodular neumocitaria multifocal (HMNM) en el 42,9% de los pacientes (5 mujeres y 4 varones). El 90,5% de los pacientes presentaba afectación renal: 16 pacientes con angiomiolipomas renales (76,2%), 12 con quistes renales (57,1%) y en 9 se observaba coexistencia de ambos (42,9%) El 28,6% presentaban función renal alterada, 1 de ellos en prediálisis. En un 38,1% se detectaron lesiones óseas escleróticas.
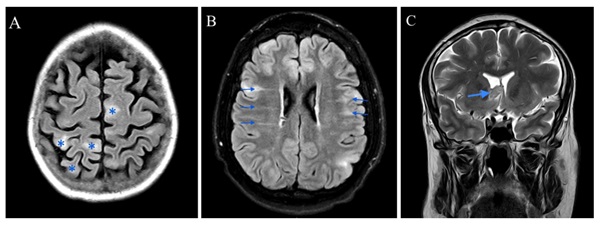
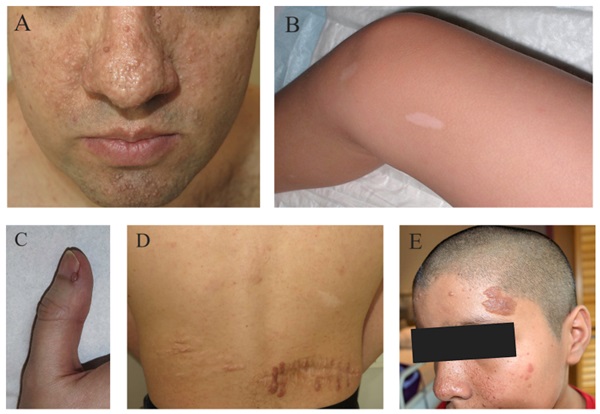
Conclusiones: Las manifestaciones de la ET más prevalentes de nuestra serie fueron las neurológicas, las dermatológicas y las renales. De cada una de ellas, las manifestaciones más frecuentes fueron los angiofibromas faciales (dermatológica); los túberes corticales y los SEN (neurológica); y los angiomioliponas renales (a nivel renal). Se describe una elevada prevalencia de LAM entre las pacientes del estudio. En ningún paciente con estudio genético positivo se detectó mutación en TSC1, siendo la totalidad de los casos detectados positivos para TSC2.


