



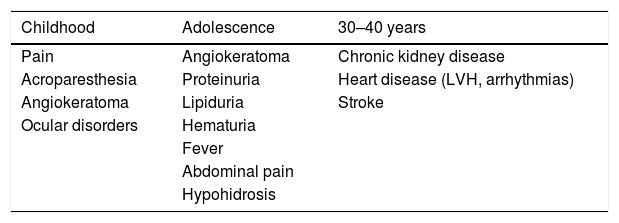
Fabry disease is an X-linked inborn disease caused by deficit of alpha-galactosidase A. This results in accumulation of glycosphingolipids in all cells and tissues. All males should receive enzyme replacement treatment in case of very low or undetectable levels of alpha-galactosidase A. Female carriers and males with marginally levels of alpha-galactosidase A should be treated in case of renal, neurologic o cardiac manifestations. There are two intravenous formulations of human recombinant enzyme, agalsidase alpha and agalsidase beta, showing similar efficacy and safety. Patients with amenable mutations of alpha-galactosidase can be treated with oral migalastat hydrochloride. Migalastat hydrochloride is a pharmacological chaperone that facilitates trafficking of alpha-galactosidase A to lysosomes increasing enzyme activity. Patients treated with migalastat hydrochloride had significant improvements in left ventricular mass and gastrointestinal symptoms.
La enfermedad de Fabry es una enfermedad ligada al cromosoma X causada por un déficit de alfa-galactosidasa A. Esto da como resultado la acumulación de glicoesfingolípidos en todas las células y tejidos. Todos los varones deben tratarse con reemplazo enzimático en caso de presentar niveles muy bajos o indetectables de alfa-galactosidasa A. Las mujeres portadoras y los varones con niveles mínimos de alfa-galactosidasa A deben tratarse si existe afectación renal, neurológica o cardíaca. Para terapia de reemplazo enzimático existen dos formulaciones intravenosas, agalsidasa alfa y agalsidasa beta, que muestran una eficacia y seguridad similares. Los pacientes con mutaciones susceptibles del gen alfa-galactosidasa A pueden tratarse con migalastat oral. El migalastat es una molécula que facilita el paso de alfa-galactosidasa A a los lisosomas, produciendo aumento de actividad del enzima. Los pacientes tratados con migalastat muestran mejoras significativas en la masa del ventrículo izquierdo y en los síntomas gastrointestinales.
Article
Diríjase desde aquí a la web de la >>>FESEMI<<< e inicie sesión mediante el formulario que se encuentra en la barra superior, pulsando sobre el candado.
Una vez autentificado, en la misma web de FESEMI, en el menú superior, elija la opción deseada.

>>>FESEMI<<<


