


660 - XANTOMATOSIS CEREBROTENDINOSA: UNA ENFERMEDAD ULTRA-RARA
Hospital Universitario Clínico San Cecilio, Granada, España.
Objetivos: La xantomatosis cerebrotendinosa (XCT) es un error congénito del metabolismo con una incidencia de 3-5/100.000 personas. De herencia autosómica recesiva, está causada por variantes patogénicas bialélicas en el gen que codifica la enzima esterol 27-hidroxilasa (CYP27A1;2q33-qter), responsable de la conversión del colesterol en ácido cólico y quenodesoxicólico (AQDC) en la vía de la síntesis de ácidos biliares, que conduce a una disminución de esta, la producción excesiva de colestanol y su acúmulo en los tejidos. Describimos las características de dos pacientes diagnosticadas en edad adulta y resaltamos los datos clínicos de sospecha al tratarse de una enfermedad ultrarrara con afectación multisistémica.
Métodos: Ambas pacientes son mujeres de 41 años, sin antecedentes familiares de interés para el caso y sin consanguinidad. Remitidas a consulta de Enfermedades Minoritarias, desde consulta de Neurología, donde realizamos el diagnóstico y su seguimiento. Se ha obtenido el consentimiento informado a las pacientes.
Resultados: Las características clínicas y el resultado del estudio genético se adjuntan en la tabla. Su perfil de ácidos biliares y esteroles séricos muestra un importante incremento de beta-colestanol y de los precursores de la síntesis de colesterol 8(9)-colestenol y 7-dehidrocolesterol, compatible con XCT. La evolución de los niveles de ácidos biliares séricos antes y después del tratamiento con AQDC se detalla en la figura. Ambas muestran una buena respuesta bioquímica al tratamiento con AQDC.
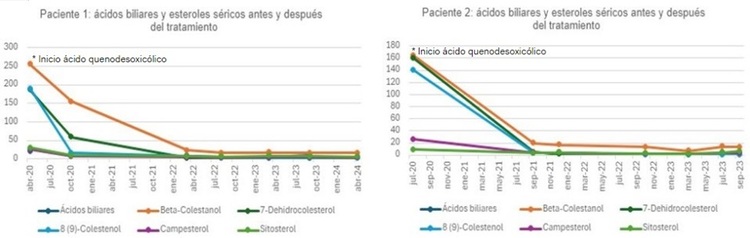
Discusión: La manifestación inicial puede ser colestasis neonatal o diarrea crónica desde la infancia, con retraso del crecimiento. Además cursa con cataratas infantiles (hasta 75% de los casos), xantomas en 2ª-3ª décadas de vida, aterosclerosis prematura, pie cavo y manifestaciones neuropsiquiátricas variables (discapacidad intelectual, demencia, trastornos psiquiátricos, piramidales y/o cerebelosos; neuropatía periférica y convulsiones). Ante su sospecha se debe solicitar determinación de ácidos biliares en sangre (siendo característica una concentración elevada de colestanol y normal-baja de colesterol) y el diagnóstico de confirmación se establece por el estudio genético. En adultos, el tratamiento de elección es el AQDC (medicamento huérfano) en dosis 250 mg tres veces al día, para síntomas neurológicos y no neurológicos como la diarrea, siendo más eficaz cuando se inicia temprano. El tratamiento con ácido cólico también es eficaz, en pacientes con efectos secundarios al AQDC. Los inhibidos de la HMG-CoA reductasa, que puede utilizarse en monoterapia o junto al AQDC, si bien puede ocasionar miopatía.
Conclusiones: Sospechar una XCT en adultos que presenten al menos dos criterios: diarrea intratable, cataratas preseniles, xantomas tendinosos y/o afectación neuropsiquiátrica, pues dispone de un tratamiento eficaz.
Bibliografía
1. Patni N, Wilson DP. Cerebrotendinous xanthomatosis (Internet). Endotext; 2023 (actualizado 8 de marzo de 2023; citado 25 mayo 2024). Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK395578/.


