


490 - ELEVACIÓN DE FACTORES DE COMPLEMENTO EN LA ENFERMEDAD DE FABRY
1Hospital Ramón y Cajal, Madrid, España. 2Universidad Alcalá de Henares, Madrid, España. 3Universidad Complutense de Madrid, Madrid, España. 4Hospital 12 Octubre, Madrid, España.
Objetivos: Estudiar la respuesta inmunológica de los pacientes con enfermedad de Fabry (FD) y compararla con un grupo control.
Métodos: Estudio observacional prospectivo transversal. Se calculó un tamaño muestral de 20 pacientes con diagnóstico de FD y 20 controles, pareados por edad (± 5 años) y sexo. Se excluyeron aquellos pacientes con enfermedades autoinmunes, autoinflamatorias, trasplantados o en tratamiento inmunosupresor, enfermedades intercurrentes graves y aquellos con evento cardiovascular agudo o cirugía mayor en los 90 días previos. Se midieron los niveles séricos de IgA, IgM, IgG, factores de complemento C3 y C4. Los resultados se expresan en mediana y rango. Se compararon lo grupos mediante la U de Mann-Whitney. Fue aprobado por el Comité Ético.
Resultados: Se incluyeron 16 pacientes con FD y 16 controles. No se alcanzó el tamaño muestral previsto por dificultades en el reclutamiento. La mediana de edad fue de 44 años en el grupo FD y 46 años en el grupo control con predominio del sexo femenino (12 en cada grupo). El 50% de los pacientes con FD recibían tratamiento específico para la enfermedad en el momento del reclutamiento. La mediana de tiempo en tratamiento era de 2 años, con un rango intercuartílico de 10 años. Los resultados de la inmunohistoquímica pueden observarse en la tabla y la figura.
Discusión: La FD es una enfermedad minoritaria, progresiva y sistémica ligada al cromosoma X. Se produce por el déficit del enzima α-galactosidasa A, que ocasiona el acúmulo de globotriaosilceramida (Gb3) en los lisosomas. Los primeros síntomas, como las acroparestesias, los angioqueratomas o los episodios febriles; pueden aparecer en la infancia. Más adelante, los pacientes clásicamente desarrollan complicaciones cardiacas, renales y cerebrales1. El daño a nivel de estos órganos parece íntimamente relacionado con un estado inflamatorio crónico mediado por el acúmulo progresivo e irreversible de Gb3 en los lisosomas. Es posible que la Gb3 actúe como patrón molecular asociado al daño (DAMP), cuya presencia a largo plazo produce una activación continua de la cascada inflamatoria. Esta activación implica al sistema del complemento y contribuye al daño tisular. Así, algunos autores proponen la contribución de procesos autoinflamatorios al desarrollo de la enfermedad2. Estudios previos mostraron niveles elevados de C4 y C3 en pacientes con FD respecto a controles y su disminución con la terapia de reemplazo enzimático3. En nuestro trabajo, la diferencia de C4 entre los dos grupos se mantiene a pesar de la inclusión de 8 pacientes con tratamiento dirigido para FD. Se observa también una tendencia a mayores niveles de C3 en el grupo con FD, que no alcanza la significación estadística, posiblemente por el pequeño tamaño de la muestra. Estos resultados apoyan la hipótesis de que los pacientes con FD presentan un estado inflamatorio crónico cuyo papel queda por definir. Los resultados expuestos son preliminares y se correlacionarán con parámetros clínicos.
|
|
Fabry (n = 16) |
Control (n = 16) |
p |
|
IgG, mg/dL |
1.100 (930-1.300) |
1.100 (750-1.600) |
0,512 |
|
IgA, mg/dL |
190,0 (144-321) |
226,5 (82,6-299) |
0,651 |
|
IgM, mg/dL |
113,5 (52,9-205) |
102,5 (40,2-214) |
0,309 |
|
Factor C3 del complemento, mg/dL |
122,5 (104-164) |
110,5 (84,3-158) |
0,086 |
|
Factor C4 del complemento, mg/dL |
33,7 (23,9-57,9) |
27,4 (16,5-42,6) |
0,021 |
|
Datos expresados en mediana (rango). |
|||
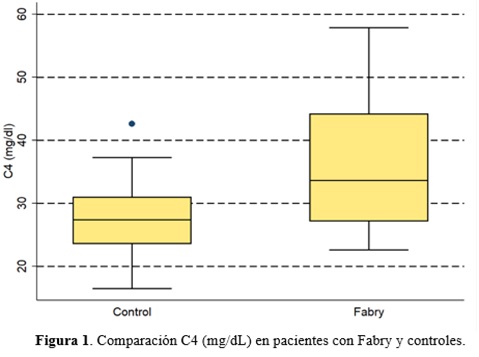
Conclusiones: Los pacientes con FD presentan una elevación de los factores C3 y C4 que podría relacionarse con la acumulación de Gb3.
Bibliografía
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30
- Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122:19-27.
- Heo SH, Kang E, Kim Y, et al. Fabry disease: characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J Med Genet. 2017;54:771-80.


