




EM-020 - AMILOIDOSIS HEREDITARIA POR TRANSTIRETINA (AHTTR). TRATAMIENTO Y PROGRESIÓN
Medicina Interna. Hospital Universitario Son Llàtzer. Palma de Mallorca (Illes Balears).
Objetivos: Analizar las diferencias en cuanto a progresión entre pacientes con AhTTR trasplantados hepáticos (TH) y pacientes en tratamiento con tafamidis.
Material y métodos: Se realiza un estudio descriptivo retrospectivo de una cohorte de pacientes con AhTTR, analizando características basales y progresión a los 6, 12 y 24m en ambos grupos.
Resultados: Se recogen en total 50 pacientes, 30 TH y 20 en tratamiento con tafamidis. De los pacientes TH 13 (43,33%) eran hombres; previo al TH 28 se encontraban en estadios 0-2 PND (3 estadio 0, 15 estadio 1 y 10 estadio 2) y en 2 el estadio era desconocido; 28 presentaban afectación neurológica (21 con electromiograma patológico), 13 disautonomía, 14 digestiva, 10 cardiaca y 9 ocular. Respecto al grupo de tafamidis el 10 (50%) eran hombres; todos estaban en estadio 1 PND y todos presentaban afectación neurológica (19 con electromiograma patológico), 14 disautonomía, 10 afectación digestiva, cardiaca (5 confirmado con gammagrafía de pirofosfato) y 2 afectación ocular. En cuanto a la progresión de ambos grupos (tabla 1). Del total de pacientes fallecieron 14 (28%), 1 en el grupo de tafamidis y 13 en TH. Se calculó el índice de masa corporal (IMC), NIS y NT-proBNP basales y post-tratamiento (tablas 2 y 3).
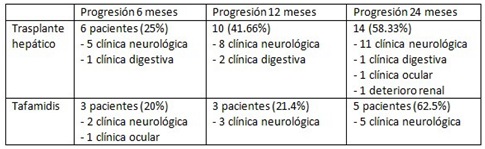
Tabla 1.
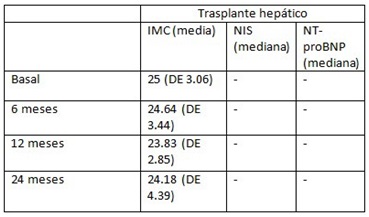
Tabla 2.
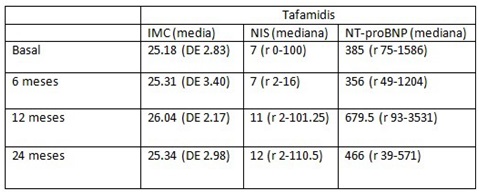
Tabla 3.
Discusión: Al comparar la evolución en ambos grupos, se observó mayor progresión en pacientes con TH que con tafamidis aunque sin diferencias estadísticamente significativas. También se observó aumento de IMC en pacientes tratados con tafamidis, tampoco significativo.
Conclusiones: La AhTTR es una enfermedad progresiva sin tratamiento curativo. El avance terapéutico con la introducción de tafamidis ha supuesto un cambio en la evolución de la enfermedad, con resultados positivos en cuanto a la reducción de progresión y mortalidad. Los datos de nuestro estudio parecen correlacionarse con los ya descritos en la bibliografía, no obstante, la posibilidad de incluir en el estudio una muestra mayor posiblemente nos permitiría encontrar resultados con significación estadística.
Bibliografía
- Ando, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet Journal of Rare Diseases. 2013;8:31.
- Conceiçao I, et al. Hereditary amyloidosis related to transthyretin V30M (hATTR V30M): disease progression in treated and untreated patients. Eur J Neurol. 2018;25(11):1320-e115.


