





Los estudios de prevalencia del déficit de esfingomielinasa ácida (ASMD) son escasos y dispares en España. El objetivo del presente estudio fue determinar la prevalencia estimada de pacientes diagnosticados de ASMD (tipos A/B y B) en España.
Material y métodosPREVASMD fue un estudio descriptivo, multicéntrico y ecológico en el que participaron 21 médicos de diferentes especialidades (principalmente de medicina interna, pediatría y hematología) de distintas comunidades autónomas, con experiencia en el manejo del ASMD.
ResultadosEntre marzo y abril de 2022 los especialistas estaban atendiendo a un total de 34 pacientes con diagnóstico de ASMD, 10 pacientes pediátricos menores de 18 años (29,4%) y 24 pacientes adultos (70,6%). La prevalencia estimada de pacientes (pediátricos y adultos) diagnosticados con ASMD era 0,7 por 1.000.000 de habitantes (intervalo de confianza 95% [IC 95%]: 0,5-1,0), 1,2 por 1.000.000 (IC 95%: 0,6-2,3) en población pediátrica y 0,6 por 1.000.000 de habitantes (IC 95%: 0,4-0,9) en población adulta. Los síntomas más frecuentes que llevaron a la sospecha del ASMD fueron: esplenomegalia (informada por el 100,0% de los especialistas), hepatomegalia (66,7%), enfermedad pulmonar intersticial (57,1%) y trombocitopenia (57,1%). Según los especialistas las pruebas de laboratorio y rutinarias, y la asistencia en atención primaria, fueron los recursos sanitarios más relevantes en el manejo del ASMD.
ConclusionesEste primer estudio realizado en España muestra una prevalencia estimada de pacientes de 0,7 por 1.000.000 de habitantes: 1,2 por 1.000.000 de habitantes en población pediátrica y 0,6 por 1.000.000 de habitantes en población adulta.
Prevalence studies of acid sphingomyelinase deficiency (ASMD) are scarce and different in Spain. The objective of the present study was to determine the estimated prevalence of patients diagnosed with ASMD (types A/B and B) in Spain.
Material and methodsPREVASMD was a descriptive, multicenter, and ecological study involving 21 physicians from different specialties (mainly Internal Medicine, Pediatrics and Hematology), of different autonomous communities, with experience in ASMD management.
ResultsBetween March and April 2022, specialists were attending a total of 34 patients with ASMD diagnosis, 10 pediatric patients under 18 years of age (29.4%) and 24 adult patients (70.6%). The estimated prevalence of patients (pediatric and adult) diagnosed with ASMD was 0.7 per 1,000,000 inhabitants (95% confidence interval, 95% CI: 0.5-1.0), 1.2 per 1,000,000 (95% CI: 0.6-2.3) in the pediatric population and 0.6 per 1,000,000 inhabitants (95% CI: 0.4-0.9) in the adult population. The most frequent symptoms that led to suspicion of ASMD were: splenomegaly (reported by 100.0% of specialists), hepatomegaly (66.7%), interstitial lung disease (57.1%), and thrombocytopenia (57.1%). According to the specialists, laboratory and routine tests, and assistance in Primary Care were the most relevant healthcare resources in the management of ASMD.
ConclusionsThis first study carried out in Spain shows an estimated prevalence of patients of 0.7 per 1,000,000 inhabitants: 1.2 per 1,000,000 inhabitants in the pediatric population and 0.6 per 1,000,000 inhabitants in the adult population.
El déficit de esfingomielinasa ácida (ASMD, del inglés Acid SphingoMyelinase Deficiency), antes conocida como enfermedad de Niemann-Pick A, o B (o NPD, por sus siglas en inglés), es una enfermedad lisosomal ultra rara, caracterizada por la acumulación de esfingomielina, especialmente en hepatocitos y células del sistema reticuloendotelial1. Los órganos más frecuentemente afectados son hígado, bazo, pulmón, médula ósea y ganglios linfáticos y, en casos más graves, el sistema nervioso central2,3.
El ASMD es una enfermedad muy desconocida, caracterizada por una gran heterogeneidad e inespecificidad de sus síntomas, lo que dificulta mucho su diagnóstico, que puede llegar a retrasarse muchos años4,5. Aunque el cuadro clínico es inespecífico, se comienza a sospechar en pacientes con hepatoesplenomegalia, trombocitopenia, enfermedad pulmonar intersticial, hiperlipidemia, retraso del desarrollo y/o mancha rojo cereza en el fondo de ojo (sobre todo en pacientes pediátricos). La confirmación diagnóstica se basa en técnicas bioquímicas y genéticas. La técnica gold standard de diagnóstico es la medición de la actividad de la enzima esfingomielinasa ácida en gota de sangre seca, que puede realizarse en linfocitos de sangre periférica o en cultivo de fibroblastos de la piel6. Tras la obtención de un diagnóstico positivo enzimático, se recomienda complementar esta confirmación con un estudio genético para detectar las variantes patogénicas del gen SMPD16,7.
La evolución de la enfermedad varía desde las formas rápidamente progresivas y fatales que presentan los niños, hasta las formas crónicas de progresión menos rápida en los adultos8. El ASMD presenta diferentes fenotipos: ASMD tipo A, tipo neurovisceral infantil; ASMD tipo B, tipo visceral crónico; y ASMD tipo A/B, tipo neurovisceral crónico9. Los tipos A y A/B se asocian con afectación neurológica. El tipo A tiene una historia natural muy uniforme, caracterizándose por hepatoesplenomegalia a los 2-4 meses de vida y una neurodegeneración progresiva desde los primeros meses de vida, con una supervivencia de menos de 3 años9. En el ASMD de tipo B, la forma más frecuente, no existe afectación neurológica (o es muy leve) y es la forma de la enfermedad que presenta una mayor variabilidad, tanto en su cuadro clínico como en la esperanza de vida, que puede llegar hasta los 70 años o concluir en una muerte temprana debido a las complicaciones relacionadas con la enfermedad. En este fenotipo predominan claramente las manifestaciones viscerales9.
Los estudios de prevalencia del ASMD son muy escasos y presentan datos dispares (entre 0,3 y 0,6 casos por cada 100.000 o 250.000 nacimientos vivos)10–14. Recientemente, unas guías clínicas de consenso para el manejo del ASMD han presentado una prevalencia de aproximadamente de 1 por 100.000 o 1.000.000 nacimientos1. Hasta el momento no se han realizado estudios epidemiológicos en pacientes con esta enfermedad en España. Por ello, el objetivo principal del presente estudio fue determinar la prevalencia estimada de pacientes diagnosticados de ASMD (tipos A/B y B) en España.
MétodosDiseño del estudioPREVASMD fue un estudio descriptivo, multicéntrico y ecológico en el que participaron 21 médicos de diferentes especialidades de distintas comunidades autónomas de España con experiencia en el manejo del ASMD. La fuente de los datos fue la experiencia de los especialistas, recogida a través de un cuestionario diseñado para tal efecto. El enfoque del estudio se basó en el análisis de la población en su conjunto (datos agregados). Por lo tanto, no se obtuvieron datos de las historias clínicas ni datos individuales de los pacientes. El estudio fue aprobado por el Comité Ético del Hospital Universitario Ramón y Cajal de Madrid.
Variables del estudio y cuestionarioLa variable principal fue la prevalencia estimada de pacientes (pediátricos y adultos) diagnosticados de ASMD, en concreto de tipo A/B y B, a partir del número de pacientes atendidos por los especialistas participantes. Otras variables secundarias analizadas fueron describir el perfil sociodemográfico y clínico de estos pacientes y la carga de la enfermedad en el sistema sanitario. El cuestionario constaba de 29 preguntas dirigidas a obtener información sobre el objetivo principal. Se recogieron, por tanto, los datos agregados obtenidos de los pacientes con ASMD (tipo A/B y B) que acudieron a la consulta del profesional sanitario con las características definidas para cada apartado del cuestionario. Adicionalmente, también se incluyeron algunas preguntas específicas que abordan los objetivos secundarios del estudio descritos anteriormente. El cuestionario se presenta en el material suplementario.
Análisis estadísticoEl análisis estadístico consistió en un análisis descriptivo de las respuestas del cuestionario. Las variables cuantitativas se muestran como media y desviación estándar (DE), mientas que las cualitativas como frecuencias absolutas y relativas. Los datos perdidos no han sido considerados en los análisis. Para el cálculo de prevalencia se consideró el dato de la población total española proporcionada por el Instituto Nacional de Estadística a fecha de 1 de enero de 2022 (47.432.805 como población total, 39.295.441 como total de población adulta y 8.137.364 como total de población pediátrica, de 0 a 17 años)15. El peso medio se estimó utilizando el promedio ponderado de la mediana de los rangos de peso de los pacientes presentados por los especialistas. Todos los análisis se han realizado utilizando el programa estadístico Statistical Package for the Social Sciences (SPSS) versión 22.0 (SPSS Inc., Chicago, IL, EE. UU.).
ResultadosEspecialistas participantesEntre marzo y abril de 2022 participaron en el estudio a un total de 21 especialistas de 9 comunidades autónomas. La lista de comunidades autónomas se muestra en la tabla 1 del material suplementario. Los especialistas tenían una experiencia clínica media de 17,6 años (DE: 8,4). Sus especialidades incluían medicina interna (38,1%), pediatría (28,6%), hematología (23,8%), gastroenterología (4,8%) y unidad metabólica (4,8%).
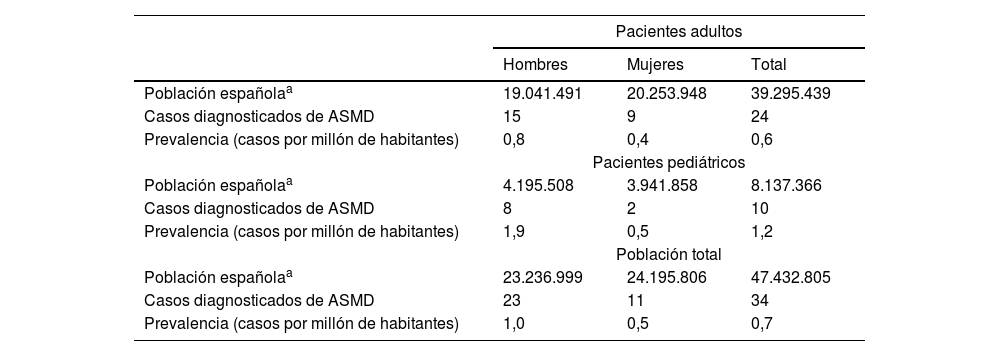
Prevalencia de déficit de esfingomielinasa ácida en EspañaLos especialistas estaban atendiendo a un total de 34 pacientes con diagnóstico del ASMD, 10 de ellos pediátricos, menores de 18 años (29,4%), y 24 pacientes adultos con edad igual o superior a 18 años (70,6%). La prevalencia estimada de pacientes (pediátricos y adultos) diagnosticados con ASMD tipo A/B y B en España fue de 0,7 por 1.000.000 habitantes (intervalo de confianza 95% [IC 95%]: 0,5-1,0) para la población total, 1,2 por 1.000.000 habitantes (IC 95%: 0,6-2,3) en población pediátrica y 0,6 por 1.000.000 habitantes (IC 95%: 0,4-0,9) en población adulta (tabla 1). La prevalencia estimada de ASMD (tipos A/B y B) en las comunidades autónomas donde se identificaron casos se presenta en la figura 1 del maerial suplementario.
Prevalencia estimada del ASMD en España
| Pacientes adultos | |||
|---|---|---|---|
| Hombres | Mujeres | Total | |
| Población españolaa | 19.041.491 | 20.253.948 | 39.295.439 |
| Casos diagnosticados de ASMD | 15 | 9 | 24 |
| Prevalencia (casos por millón de habitantes) | 0,8 | 0,4 | 0,6 |
| Pacientes pediátricos | |||
| Población españolaa | 4.195.508 | 3.941.858 | 8.137.366 |
| Casos diagnosticados de ASMD | 8 | 2 | 10 |
| Prevalencia (casos por millón de habitantes) | 1,9 | 0,5 | 1,2 |
| Población total | |||
| Población españolaa | 23.236.999 | 24.195.806 | 47.432.805 |
| Casos diagnosticados de ASMD | 23 | 11 | 34 |
| Prevalencia (casos por millón de habitantes) | 1,0 | 0,5 | 0,7 |
ASMD: déficit de esfingomielinasa ácida.
La mayoría de pacientes con diagnóstico del ASMD eran hombres (67,7%) y adultos (70,6%). Las características sociodemográficas y clínicas de los pacientes con diagnóstico de ASMD se muestran en la tabla 2. El peso medio estimado de los pacientes pediátricos (<18años) fue 29,4kg, mientras que el de los adultos (≥18 años) fue 70,5kg. El tiempo medio desde el inicio de los síntomas (no específicos) hasta el diagnóstico del ASMD fue de 2,5 años (DE: 3,5) y desde el diagnóstico hasta el inicio del tratamiento sintomático fue de 2,3 años (DE: 4,2). Los síntomas más comunes que presentaron los pacientes con ASMD fueron: esplenomegalia (indicada por el 90,5% de los especialistas), hiperlipidemia (66,7%), enfermedad pulmonar intersticial (57,1%), hepatomegalia (57,1%) y trombocitopenia (52,4%). De entre esos síntomas, los que incitaron al especialista a sospechar del ASMD fueron: esplenomegalia (100,0%), hepatomegalia (66,7%), enfermedad pulmonar intersticial (57,1%) y trombocitopenia (57,1%; fig. 1A). Según los especialistas el pronóstico y la progresión de la enfermedad estuvo determinado principalmente por la gravedad de la afectación pulmonar (100,0% de ellos; n disponible=21), seguido por la gravedad de la afectación hepática (81,0%), la edad del paciente (52,4%), la actividad enzimática residual (52,4%) y factores genéticos (42,9%; fig. 1B).
Características sociodemográficas y clínicas de los pacientes con diagnóstico del ASMD (de acuerdo a los especialistas participantes)
| Pacientes (n=34) | |
|---|---|
| Hombres, n (%) | 23 (67,7) |
| Rango de edad, n (%) | |
| 0-6 | 2 (5,9) |
| 6-12 | 4 (11,8) |
| 12-17 | 4 (11,8) |
| 18-39 | 12 (35,3) |
| 40-59 | 6 (17,6) |
| 60-89 | 6 (17,6) |
| Tiempo desde el inicio de los síntomas (no específicos) hasta el diagnóstico de ASMD, media (DE) años | 2,5 (3,5) |
| Tiempo desde el diagnóstico de ASMD hasta el inicio del tratamiento sintomático, media (DE) años | 2,3 (4,2) |
ASMD: déficit de esfingomielinasa ácida; DE: desviación estándar.
 y factores que determinan el pronóstico y curso de la enfermedad (B).")
A falta de la existencia de un tratamiento enzimático (aprobado por la Agencia Europea de Medicamentos, pero sin precio en España), en opinión de los especialistas el 66,7% de los pacientes estaba recibiendo tratamientos sintomáticos, el 19,0% fisioterapia y el 19,0% oxigenoterapia (fig. 2A). Los tratamientos sintomáticos más frecuentes fueron los hipolipidemiantes (33,3%) y los analgésicos (28,6%; fig. 2B). El 76,2% de los especialistas indicaron haber solicitado pruebas enzimáticas/genéticas para el diagnóstico de la enfermedad. En los casos que no se solicitaron pruebas enzimáticas/genéticas el diagnóstico se alcanzó entrelazando antecedentes familiares, clínica compatible/muy sugestiva de la enfermedad o estudios histológicos (especialmente biopsia de médula ósea), a la espera de los resultados enzimáticos y genéticos. Según los especialistas, los recursos sanitarios más relevantes en el manejo del ASMD fueron las pruebas de laboratorio y de rutina (24,3%) y la asistencia en atención primaria (21,1%; fig. 3A). Los especialistas indicaron que principalmente utilizaban las visitas médicas (100,0% de ellos), análisis de sangre (100,0%), ecografías (95,2%), pruebas de difusión pulmonar (85,7%) y radiografías de tórax (85,7%) para el seguimiento de sus pacientes (fig. 3B). El 19,0% de los especialistas informaron de que sus pacientes fueron hospitalizados una vez al año de promedio. Las principales causas de hospitalización de los pacientes con ASMD eran las insuficiencias respiratorias (38,1%), la neumonía (23,8%) y las insuficiencias hepáticas (9,5%). El 28,6% de los especialistas indicó que sus pacientes acudieron a urgencias por su enfermedad entre una y 4 veces durante el último año. Finalmente, señalaron que ninguno de sus pacientes requirió una intervención quirúrgica.
 y, entre ellos, los sintomáticos (B) prescritos a pacientes con diagnóstico de déficit de esfingomielinasa ácida. AUDC, ácido ursodesoxicólico; PPVR: presión positiva continua en las vías respiratorias.")
 y pruebas rutinarias y seguimiento clínico (B) en pacientes con diagnóstico de déficit de esfingomielinasa ácida. * Otras incluye densitometría ósea, polisomnografía, resonancia magnética pulmonar, resonancia magnética torácica, ecocardiografía, espirometría y la prueba de capacidad de difusión de monóxido de carbono.")
Recursos sanitarios más empleados (A) y pruebas rutinarias y seguimiento clínico (B) en pacientes con diagnóstico de déficit de esfingomielinasa ácida.
* Otras incluye densitometría ósea, polisomnografía, resonancia magnética pulmonar, resonancia magnética torácica, ecocardiografía, espirometría y la prueba de capacidad de difusión de monóxido de carbono.
El presente trabajo es el primer estudio epidemiológico del ASMD en España, presentando una prevalencia estimada de 0,7 casos por 1.000.000 de habitantes, que es mayor en la población pediátrica (1,2) que en la adulta (0,6). Estudios previos realizados en diferentes países (República Checa, Países Bajos, Portugal y Emiratos Árabes Unidos) han mostrado cifras de prevalencia conjunta al nacer de ASMD tipos A y B entre 0,3 y 0,6 casos por cada 100.000 nacimientos vivos11–14. Sin embargo, un estudio previo en Australia presentó un valor de 1 por cada 248.000 nacimientos vivos10. En nuestro estudio, la prevalencia conjunta de ASMD tipos A/B y B, determinada en población pediátrica y adulta, se encuentra por debajo de los estudios publicados previamente4. En nuestra opinión, diferencias metodológicas en la determinación de la prevalencia podrían explicar tales discrepancias. Por un lado, los estudios previos evaluaron la prevalencia al nacer, por lo que el cálculo se realizó basándose en el número total de casos diagnosticados que habían nacido dentro de un cierto período de tiempo respecto al número de nacimientos en ese mismo período. Además, estos estudios determinaron la prevalencia de manera conjunta entre ASMD tipos A y B, a diferencia de nuestro estudio, que fueron los tipos A/B y B. En cualquier caso, todas las estimaciones de prevalencia, tanto en la literatura11–14 como en nuestro estudio, se basaron en casos sospechosos que fueron remitidos por especialistas para su confirmación bioquímica y/o molecular, y no en estudios de cribado en población general. El único estudio de cribado disponible se realizó en Chile sobre 1.691 individuos sanos en los que determinó una de las variantes patogénicas del ASMD (p.Ala359Asp)16. Este estudio registró una incidencia de la enfermedad de un caso por 44.960 individuos. Dada la limitación de información disponible, se considera que el ASMD está muy infradiagnosticado en la práctica clínica habitual, especialmente en los casos de ASMD tipo B, donde el diagnóstico clínico puede no ser fácilmente evidente.
En este sentido, las manifestaciones clínicas del ASMD pueden superponerse con otras enfermedades de depósito lisosomal, como la enfermedad de Gaucher10. Entre las manifestaciones clínicas, la hepatoesplenomegalia es una de las más frecuentes para su identificación17. La presencia de enfermedad pulmonar intersticial y dislipidemia puede ayudar a distinguir el ASMD de otras enfermedades, como las neoplasias malignas hematológicas, que pueden presentarse con hepatoesplenomegalia y pancitopenia10. No obstante, la confirmación diagnóstica definitiva del ASMD se realiza mediante pruebas bioquímicas y/o genéticas18,19. En nuestro estudio, los especialistas identificaron como síntomas principales que conducen al diagnóstico de la enfermedad la esplenomegalia, la hepatomegalia, la enfermedad pulmonar intersticial y la trombocitopenia. Es importante además resaltar que los especialistas asociaron especialmente la gravedad de la afectación pulmonar y hepática como factores principales que determinan el pronóstico y curso de la enfermedad. Como se ha presentado en estudios previos, existe un desfase significativo entre la presentación inicial de los síntomas y el diagnóstico definitivo de la enfermedad (aproximadamente 5 años de media)20. En nuestro estudio el retraso diagnóstico de la enfermedad fue ligeramente inferior (2,5 años). Respecto a los pacientes con ASMD es necesario hacer hincapié en que, en nuestro estudio, estos fueron mayoritariamente adultos (70,6%). Por el contrario, otros estudios muestran datos similares o mayoritariamente pediátricos20,21. Por ejemplo, en un estudio multicéntrico, multinacional de encuesta transversal, con datos de 59 pacientes con ASMD tipo B, el 50,8% de estos eran pediátricos20. Sin embargo, un estudio con datos de 64 casos de ASMD tipo B mostró que el porcentaje de pacientes pediátricos (57,8%) era ligeramente mayor que el de adultos (42,2%)21.
En España actualmente se está llevando a cabo el proyecto PREDIGA, cuyo objetivo es identificar pacientes con enfermedad del ASMD y enfermedad de Gaucher no diagnosticados que puedan beneficiarse de tratamientos efectivos22. El proyecto, además, cuenta con un programa nacional de educación, cuya finalidad es mejorar el conocimiento de estas enfermedades y la difusión de algoritmos de diagnóstico. En el primer año del proyecto educativo, con la participación de 52 profesionales sanitarios de 34 hospitales de España, se revisaron 166 pacientes con criterios diagnósticos y alcanzaron la confirmación diagnóstica de ASMD en 2 de ellos.
Por otro lado, el ASMD se asocia con una importante carga física, emocional, psicosocial y económica sobre los pacientes y sus cuidadores23,24. Estudios previos han revelado que los pacientes con enfermedad crónica requieren atención ambulatoria que incluye fisioterapia o atención médica domiciliaria. Además, se ha comunicado que la utilización de los servicios de atención sanitaria por parte de pacientes con ASMD es amplia y puede incluir evaluaciones de laboratorio (enzimas hepáticas y paneles de lípidos), pruebas de función pulmonar, histopatología, ecografías/radiografías, tratamientos farmacológicos profilácticos y sintomáticos. También requieren procedimientos médicos, como oxigenoterapia, fisioterapia y trasplante de órganos25,26. En nuestro estudio los recursos sanitarios más empleados eran las pruebas de laboratorio y la atención primaria. Para el seguimiento de los pacientes los especialistas principalmente utilizaban las visitas médicas, análisis de sangre, ecografías, pruebas de difusión pulmonar y radiografías de tórax.
La principal limitación del presente estudio radica en la naturaleza subjetiva de los datos, proporcionados como datos agregados sobre la experiencia de los especialistas, es decir, sin incluir datos recopilados de las historias clínicas. Además, los datos pueden estar influidos por un sesgo de recuerdo. Pese a ello, los estudios ecológicos forman parte de los diseños observacionales en epidemiología y se emplean como generadores de hipótesis, al ser sencillos de realizar, rápidos, poco costosos y su análisis relativamente sencillo27. Otra limitación del estudio puede derivar del 23,8% de pacientes con ASMD, cuyo diagnóstico no pudo ser confirmado mediante estudio genético al cerrarse la base de datos. No obstante, algoritmos empleados en práctica clínica permiten establecer un diagnóstico de ASMD en función de antecedentes familiares, clínica compatible/muy sugestiva de la enfermedad o estudios histológicos. En conjunto, nuestros resultados proporcionan un primer paso de información sobre la epidemiología de la enfermedad en España, así como del perfil sociodemográfico y clínico de estos pacientes y la carga de enfermedad en el sistema sanitario.
ConclusiónEste primer estudio sobre el ASMD realizado en España muestra una prevalencia estimada de pacientes con ASMD de tipo A/B y B de 0,7 por 1.000.000 de habitantes, que es mayor en población pediátrica (1,2) que en adulta (0,6). Dados los escasos estudios epidemiológicos en esta enfermedad, se considera que está infradiagnosticada en la práctica clínica habitual. Se requieren más estudios, con mayor número de participantes e, incluso, estudios de cribado en población sana para establecer un valor lo más cercano a la realidad.
FinanciaciónEste trabajo ha sido financiado por Sanofi España. La asistencia en la redacción del manuscrito ha sido proporcionada por Evidenze Health España S.L.U., que fue contratada por Sanofi España. Sanofi España no tuvo relación directa con los autores. Ninguno de los autores ha recibido financiación directa de la industria.
Conflicto de interesesTodos los autores han mantenido independencia editorial y de opinión durante la preparación de este manuscrito y todos ellos (J. Villarrubia, M. Morales, L. Ceberio, I. Vitoria, M. Bellusci, I. Quiñones, L. Peña, M. Ruiz de Valbuena y M. O’Callaghan) declaran no tener conflicto de intereses.
Beatriz Buno Ramilo (Hospital Arquitecto Marcide, A Coruña), Alejandro Contento (Hospital Carlos Haya, Málaga), Iván Pérez de Pedro (Hospital Carlos Haya, Málaga), Lucía Villalón Blanco (Hospital Universitario Fundación Alcorcón, Madrid), Cecilia Muñoz Delgado (Hospital General Universitario Gregorio Marañón, Madrid), María del Carmen Mendoza Sánchez (Hospital Universitario de Salamanca, Salamanca), Antonio González-Meneses (Hospital Universitario Virgen del Rocío, Sevilla), Dolores Gómez Toboso (Hospital de Manises, Valencia), Moisés de Vicente (Hospital General Nuestra Señora del Prado, Toledo), Alicia Rodríguez (Hospital Universitario Virgen Macarena, Sevilla), Jose Antonio Pérez de León (Hospital Universitario Virgen Macarena, Sevilla) y Xavier Solanich (Hospital Universitario de Bellvitge, Barcelona).


