



Mujer de 32 años, sin antecedentes epidemiológicos relevantes salvo contacto tuberculoso en la infancia, remitida desde el Servicio de Oftalmología para valorar estudio de enfermedad sistémica. Refiere miodesopsias y disminución de agudeza visual en ambos ojos de varios meses de evolución, acompañados de artralgias y tos seca, sin otra sintomatología extraocular. La exploración oftalmológica muestra agudeza visual de 0,3 en el ojo derecho y 0,4 en el ojo izquierdo. En la exploración de la cámara anterior se observa una uveítis anterior bilateral granulomatosa con discreta inflamación (Tyndall 1+). Se observa vitritis (3+) bilateral, con exudados en banco de nieve y flebitis periférica. La angiografía con fluoresceína no muestra signos de vasculitis central, y la tomografía de coherencia óptica confirma la existencia de edema macular quístico bilateral. La autofluorescencia es normal. ¿Cómo debe ser evaluada inicialmente esta paciente para decidir cuál es el estudio sistémico más adecuado teniendo en cuenta las manifestaciones oftalmológicas?
A 32 year-old woman was referred from the Ophthalmology Department to rule out a possible systemic disease. Her only past medical history of relevance was a tuberculosis contact during childhood. She complained of floaters and progressive blurring of vision in both eyes for some months, as well as arthralgia and cough. Her visual acuity was 0.3 in the right eye and 0.4 in the left eye. Biomicroscopy showed bilateral anterior granulomatous uveitis (1+ cells). Funduscopy showed bilateral vitritis 3+, snow banking and peripheral phlebitis. Fluorescein angiography did not show central vasculitis, and optical coherence tomography showed bilateral cystoid macular oedema. Fundus autofluorescence was normal. How would you initially assess this patient in order to decide which systemic examination should be performed, bearing in mind the ophthalmological manifestations?
Las uveítis son un grupo heterogéneo de entidades clínicas de etiología muy variada, que causan inflamación intraocular. Aunque en sentido estricto se define como uveítis al proceso inflamatorio que afecta a la úvea, en la práctica clínica el término se emplea de forma más amplia, abarcando al conjunto de enfermedades que afectan tanto al tracto uveal como a las estructuras intraoculares adyacentes (vítreo, retina, nervio óptico y vasos). Pueden aparecer en cualquier edad, aunque son más frecuentes en la segunda, tercera y cuarta décadas de la vida, y se estima que en los países desarrollados su incidencia oscila entre 17-52 casos por 100.000 habitantes y año, con una prevalencia de 38-714 casos. Las uveítis son responsables aproximadamente del 10-15% de los nuevos casos de ceguera; se estima que alrededor del 35% de los pacientes con uveítis presenta baja visión o ceguera legal1–6.
Las uveítis pueden estar originadas por múltiples causas, por lo que la aproximación diagnóstica supone un verdadero reto tanto para el oftalmólogo como para el internista. Aunque la etiología es variada (tabla 1), podemos distinguir 4 grandes grupos de uveítis en función de su etiología: uveítis infecciosas, uveítis no infecciosas puramente oftalmológicas (uveítis idiopáticas y síndromes oftalmológicos específicos), uveítis asociadas a enfermedades autoinmunes/inflamatorias sistémicas y síndromes de enmascaramiento (procesos no inflamatorios, fundamentalmente neoplásicos, que pueden simular una uveítis). Resulta evidente la importancia de llegar a un diagnóstico etiológico concreto, que oriente el pronóstico, facilite la elección del tratamiento específico adecuado (evitando iatrogenias, especialmente en el caso de uveítis infecciosas) y permita en algunos casos el diagnóstico precoz de enfermedades sistémicas. El consenso actual recomienda que el estudio diagnóstico se individualice en función del patrón clínico de la uveítis y de las características clínicas y epidemiológicas del paciente6–8.
Etiología de las uveítis
| Uveítis infecciosas |
| – Virales: virus herpes simple, citomegalovirus, virus varicela-zóster, virus de Epstein-Barr, VIH, rubéola, parotiditis |
| – Bacterianas: tuberculosis, sífilis, enfermedad de Lyme, enfermedad por arañazo de gato, Propionibacterium, leptospirosis, rickettisiosis, enfermedad de Whipple, lepra, micobacterias atípicas |
| – Parasitarias: Toxoplasma, Toxocara, Acanthamoeba, cisticercosis, Onchocercus |
| – Fúngicas: candidiasis, aspergilosis, histoplasmosis, blastomicosis, Fusarium, criptococosis, coccidiomicosis, esporotricosis |
| Uveítis no infecciosas puramente oftalmológicas |
| – Uveítis idiopáticas: asociadas a HLA B27, no asociadas a HLA B27 |
| – Síndromes oftalmológicos bien definidos: |
| • Uveítis anteriores: ciclitis heterocrómica de Fuchs, síndrome de Posner-Schlossman, uveítis facogénicas, uveítis asociadas a lentes intraoculares |
| • Uveítis intermedias: pars planitis |
| • Uveítis posteriores y panuveítis: coroidopatía serpinginosa, coroidopatía en perdigonada, epiteliopatía pigmentaria placoide multifocal, coroiditis multifocal con panuveítis, coroidopatía punteada interna, síndrome de los puntos blancos múltiples evanescentes, epitelitis pigmentaria retiniana aguda, retinopatía externa zonal aguda, síndrome de fibrosis subretiniana y uveítis, síndrome de coroiditis multifocal y panuveítis, oftalmía simpática |
| Uveítis asociadas a enfermedades sistémicas de etiología autoinmune-inflamatoria |
| – Espondiloartropatías: espondilitis anquilosante, síndrome de Reiter y artritis reactivas, artropatía psoriásica |
| – Enfermedad inflamatoria intestinal: enfermedad de Crohn, colitis ulcerosa |
| – Artritis idiopática juvenil |
| – Sarcoidosis |
| – Enfermedad de Behçet |
| – Enfermedad de Vogt-Koyanagi-Harada |
| – Esclerosis múltiple |
| – Vasculitis sistémicas: vasculitis asociadas a ANCA, poliarteritis nodosa, enfermedad de Kawasaki, arteritis de Takayasu |
| – Conectivopatías: lupus eritematoso sistémico, síndrome de Sjögren, polimiositis, dermatomiositis |
| – Policondritis recidivante |
| – Síndrome de nefritis tubulointersticial y uveítis (TINU) |
| – Uveítis inducida por fármacos y reacciones de hipersensibilidad |
| Síndromes de enmascaramiento |
| – Neoplásicos: linfoma, leucemia, melanoma de úvea, metástasis, retinoblastoma |
| – Vasculares: síndrome antifosfolipídico (enfermedad venooclusiva) |
| – Oftalmológicos: retinitis pigmentosa, desprendimiento de retina regmatógeno periférico crónico, síndrome de dispersión pigmentaria |
ANCA: anticuerpos anticitoplasma de los neutrófilos.
La identificación de un patrón clínico lo más preciso posible es el primer paso para poder plantear adecuadamente el diagnóstico diferencial de un paciente con uveítis, y decidir qué pruebas diagnósticas es necesario realizar6–8. Para ello, es preciso integrar diferentes formas de clasificar las uveítis en función de criterios anatómicos (porción afectada de la úvea), de evolución clínica (aguda, crónica o recurrente; uni o bilateral) e histopatológicos (granulomatosa o no granulomatosa). La identificación del patrón clínico va a ser labor fundamentalmente del oftalmólogo, por lo que es será necesario familiarizarse con las técnicas diagnósticas y la terminología empleadas en la exploración oftalmológica. Desde un punto de vista práctico, tendremos que intentar responder a 4 preguntas:
¿Dónde se localiza la inflamación ocular?Probablemente, la localización anatómica de la inflamación ocular sea el dato más relevante de la exploración oftalmológica a la hora de establecer el diagnóstico de las uveítis. La clasificación habitualmente empleada se basa en las recomendaciones del grupo SUN (The Standardization of Uveitis Nomenclature Working Group) y del grupo IUSG (International Uveitis Study Group), y distingue 4 tipos de uveítis: anterior, intermedia, posterior y panuveítis9,10.
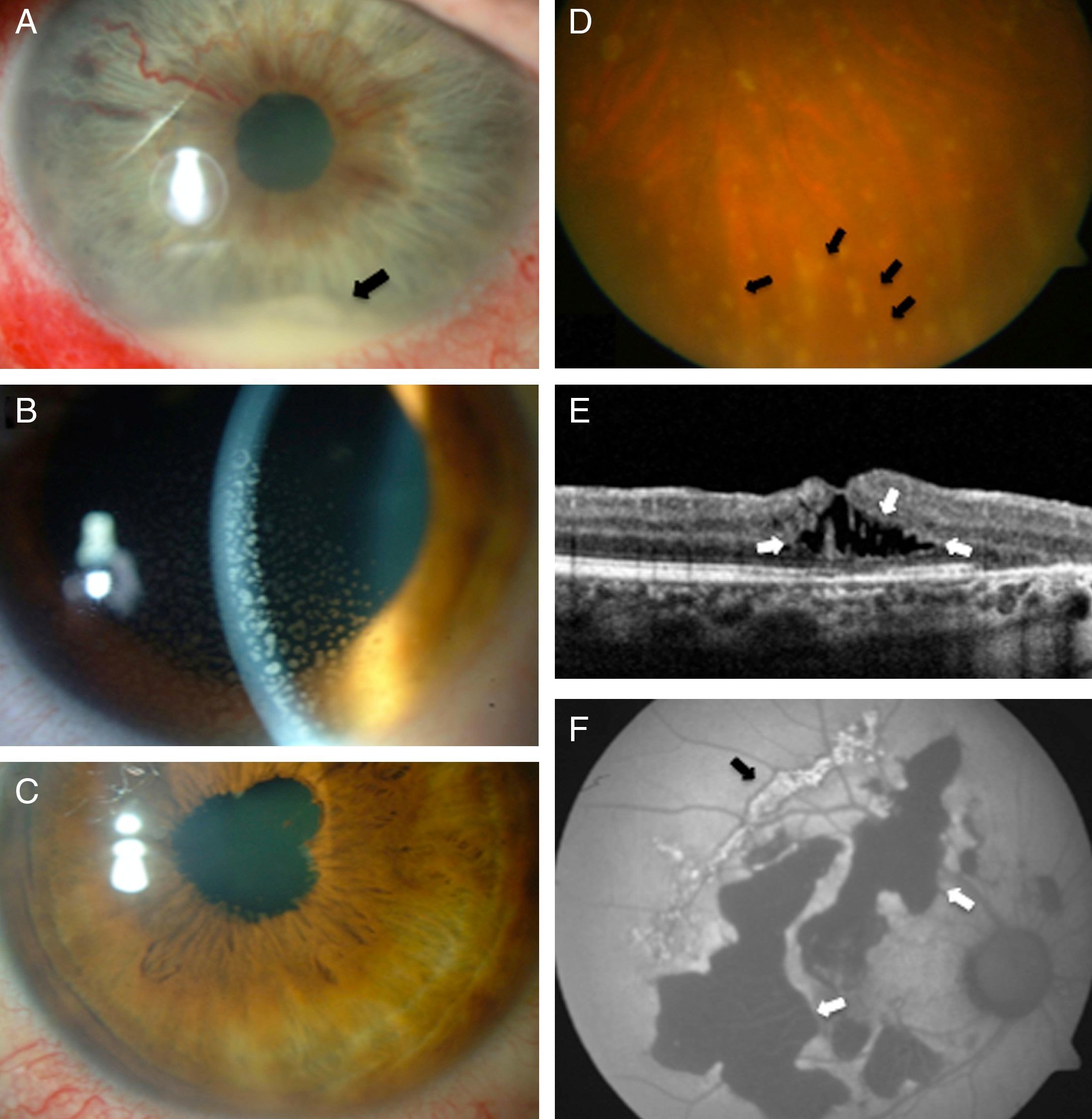
En la uveítis anterior la inflamación se localiza primariamente en la cámara anterior (iritis, iridociclitis)8–10. En ocasiones la uveítis se asocia a inflamación corneal (queratouveítis) o de la esclera (esclerouveítis). El cuadro clínico puede ir desde una inflamación leve, solo visible en la exploración detallada, hasta un cuadro con inflamación moderada o grave caracterizado por dolor, fotofobia, enrojecimiento ocular, lagrimeo y disminución variable de la agudeza visual; en ocasiones se produce elevación de la presión intraocular (PIO). En la exploración oftalmológica se observa hiperemia conjuntival y ciliar periquerática, disfunción del reflejo pupilar a los cambios luminosos y turbidez en la cámara anterior (fenómeno Tyndall); hablamos de hipopión cuando la inflamación es muy intensa y se acumulan fibrina y células inflamatorias en forma de nivel de color blanquecino en la parte inferior de la cámara anterior (fig. 1A). Pueden encontrarse también precipitados queráticos (acúmulos de células inflamatorias adheridas al endotelio corneal) y sinequias, tanto anteriores (adherencias entre la parte anterior del iris y el endotelio corneal) como posteriores (entre la parte posterior del iris y el cristalino) (fig. 1B y C).
![A) Hipopión. B) Imagen biomicroscópica de un caso de uveítis anterior herpética; el haz de hendidura está enfocado sobre el endotelio corneal donde se observan precipitados queráticos gruesos. C) Deformidad pupilar causada por la presencia de sinequias posteriores. D) Snowballs en un caso de uveítis intermedia (flechas). E) Imagen de tomografía de coherencia óptica (Optical Coherence Tomography [OCT]) que muestra edema macular (flechas). F) Imagen de autofluorescencia de un caso de coroidopatía serpinginosa, en la que se observan lesiones hiper (flecha negra) e hipofluorescentes (flecha blanca).](https://static.elsevier.es/multimedia/00142565/0000021200000009/v1_201302081208/S0014256511005856/v1_201302081208/es/main.assets/gr1.jpeg?xkr=FGFA6X66eBGksCdreAKW45UFAsvq79ERH+7yEhIOjEJ2Cm/HVAKRxWC92Jt63IPGMButpfzFmKbkX+Uj9ozAGMHb5kPKBReH25TXzchlqwy+8oiMo/Jpm6aoiOYM93CY69BoEPw6n4/D4ath8gAWocnzMVGkiYohC2cVgGZ7FbMvYyUuOsl+WrE4kwumWhXGx018+JZvuBjbN6QZQZ93seJy0TJ56271w+dMlkkVZOKWD9K+0Y+wWbSrCrw2i6Piwg2IO6Irs4LncdvfDpHBMxHnnChCyInp84pX4mHYACA= "A) Hipopión. B) Imagen biomicroscópica de un caso de uveítis anterior herpética; el haz de hendidura está enfocado sobre el endotelio corneal donde se observan precipitados queráticos gruesos. C) Deformidad pupilar causada por la presencia de sinequias posteriores. D) Snowballs en un caso de uveítis intermedia (flechas). E) Imagen de tomografía de coherencia óptica (Optical Coherence Tomography [OCT]) que muestra edema macular (flechas). F) Imagen de autofluorescencia de un caso de coroidopatía serpinginosa, en la que se observan lesiones hiper (flecha negra) e hipofluorescentes (flecha blanca).")
A) Hipopión. B) Imagen biomicroscópica de un caso de uveítis anterior herpética; el haz de hendidura está enfocado sobre el endotelio corneal donde se observan precipitados queráticos gruesos. C) Deformidad pupilar causada por la presencia de sinequias posteriores. D) Snowballs en un caso de uveítis intermedia (flechas). E) Imagen de tomografía de coherencia óptica (Optical Coherence Tomography [OCT]) que muestra edema macular (flechas). F) Imagen de autofluorescencia de un caso de coroidopatía serpinginosa, en la que se observan lesiones hiper (flecha negra) e hipofluorescentes (flecha blanca).
En la uveítis intermedia el lugar primario de inflamación es el vítreo8–10. La presentación más frecuentes suele consistir en miodesopsias (moscas volantes) y visión borrosa. El hallazgo fundamental en la exploración es la vitritis, y con frecuencia encontraremos un infiltrado inflamatorio celular vítreo en «bolas de nieve» (snowballs, fig. 1D), exudados en «banco de nieve» en la pars plana (snowbanks), flebitis retiniana periférica, papilitis (hiperemia papilar en los casos más leves) y edema macular quístico; no suele haber signos externos de inflamación ocular. Se reserva el término pars planitis para las uveítis intermedias idiopáticas con formación de snowbanks o snowballs.
En las uveítis posteriores la inflamación se localiza primariamente en la coroides (coroiditis), la retina (retinitis) o en ambos (coriorretinitis o retinocoroiditis, según se afecte inicialmente una u otra); en aquellos casos con afectación de la cabeza del nervio óptico y la retina adyacente hablaremos de neurorretinitis8–10. La coroiditis puede ser focal, multifocal o difusa. En ocasiones se observa inflamación vítrea, bien como vitritis posterior o por la presencia de células inflamatorias vítreas por encima de los focos de inflamación activa. Otros hallazgos importantes son el edema macular quístico, el desprendimiento exudativo de retina, la necrosis retiniana y los signos de vasculitis retiniana (envainamiento perivascular, oclusión vascular, hemorragias y exudados). Las manifestaciones clínicas iniciales de las uveítis posteriores son las miodesopsias y grados variables de disminución de agudeza visual.
Las panuveítis son uveítis en las que la inflamación intraocular es difusa, localizada en cámara anterior, vítreo y coroides y/o retina, sin que predomine en ningún segmento8–10. Las manifestaciones clínicas pueden ser muy variadas, con síntomas propios de los tres tipos de uveítis anteriormente descritos. La presencia de coroiditis o coriorretinitis, vitritis, vasculitis retiniana o desprendimiento exudativo de retina son de gran utilidad a la hora de establecer el diagnóstico diferencial de las panuveítis.
¿Es una uveítis granulomatosa o no granulomatosa?En las uveítis anteriores las células inflamatorias se adhieren al endotelio corneal, dando lugar a los precipitados queráticos, que pueden diferenciarse mediante la exploración con lámpara de hendidura, sin necesidad de biopsia, en no granulomatosos (pequeños, redondeados y de coloración blanquecina; constituyen un hallazgo inespecífico y poco orientativo) y granulomatosos (más grandes, de coloración con frecuencia amarillenta o marronácea, por lo que suelen denominarse «precipitados queráticos en grasa de carnero»)8. Los precipitados queráticos granulomatosos se asocian con ciertos procesos, generalmente de inicio insidioso y curso crónico (sarcoidosis, oftalmía simpática, síndrome de Vogt-Koyanagi-Harada, esclerosis múltiple, tuberculosis, sífilis, uveítis herpéticas), por lo que se trata de un hallazgo muy útil para orientar el diagnóstico. Otros hallazgos sugerentes de inflamación granulomatosa ocular son los nódulos en iris y los granulomas conjuntivales, que suelen orientar hacia ciertas etiologías (sarcoidosis, sífilis) y son excepcionales en otras uveítis (uveítis herpética, oftalmía simpática).
¿Es una uveítis aguda o crónica?Según las definiciones propuestas por el grupo SUN9, las uveítis agudas se definen por un inicio brusco y una duración limitada (inferior a tres meses con tratamiento adecuado); se denominan recurrentes cuando se producen episodios repetidos separados por períodos de inactividad sin tratamiento de más de tres meses. Las uveítis crónicas son aquellas que duran más de tres meses o que recidivan en menos de tres meses tras retirar el tratamiento; habitualmente se inician de forma insidiosa.
¿Es una uveítis unilateral o bilateral?Las uveítis unilaterales afectan a un único ojo; en las uveítis agudas recurrentes, se consideran unilaterales si se inflama un único ojo en cada brote, aunque en brotes sucesivos puede inflamarse el otro ojo (no simultáneamente ambos ojos en el mismo brote). Aunque inicialmente la uveítis puede ser unilateral, en la mayor parte de los casos acaba apareciendo en ambos ojos (de forma alterna, sucesiva o simultánea). Por ello, resulta muy interesante para el diagnóstico diferencial que la uveítis afecte a un solo ojo y respete el otro, orientando con frecuencia a una etiología infecciosa.
La exploración oftalmológica en uveítisDesde el punto de vista de un internista, la aproximación diagnóstica a las uveítis tiene la dificultad añadida que supone la interpretación de la exploración oftalmológica, exploración que realiza otro especialista, con el empleo de una terminología propia y de una serie de pruebas diagnósticas con las que con frecuencia estamos poco familiarizados. La exploración del ojo en las uveítis va encaminada a valorar tanto el tipo y grado de daño estructural como la repercusión funcional de dicho daño. Existen una serie de exploraciones básicas (aquellas que se realizan de rutina y no requieren un aparataje especial ni sofisticado) y de exploraciones complementarias (se llevan a cabo en situaciones determinadas y con indicaciones específicas) con las que conviene estar familiarizado.
Exploraciones básicasValoración funcionalLa exploración funcional más importante en oftalmología es la medida de la agudeza visual (AV) mediante optotipos de letras o números que pueden colocarse a distancias variables del paciente (habitualmente 4 metros). El resultado se expresa en forma de cociente (20/100) o decimal (0,3). Por debajo de 0,05 cuantificamos la AV en cuenta dedos, movimiento de manos, percepción de luz y no percepción de luz. Siempre debe medirse la AV con la refracción adecuada del paciente.
Valoración estructuralLa exploración oftalmológica permite observar directamente la alteración estructural del ojo. El biomicroscopio (BMC) o lámpara de hendidura permite explorar las alteraciones del segmento anterior: afección corneal, precipitados queráticos, presencia de células o fibrina en cámara anterior, alteraciones en el iris (sinequias, atrofia, nódulos) y el estado del cristalino (fig. 1B). A través de una lente que se interpone entre el haz de luz y el ojo observamos las alteraciones presentes en la cavidad vítrea y las estructuras del fondo de ojo: nervio óptico, vasos y mácula. El BMC tiene incorporado un tonómetro de aplanación (tonómetro de Goldmann) para medir la PIO. La exploración suele complementarse con el oftalmoscopio indirecto, que permite visualizar mejor la periferia retiniana. Existen dos escalas estandarizadas para cuantificar la intensidad de la inflamación ocular: para la cámara anterior, la clasificación establecida por el grupo SUN (basada en el número de células observadas por mm2 de haz de hendidura), y para la cámara vítrea la escala de Nussenblatt (basada en el grado de opacidad que produce la vitritis)8,9. No existe ninguna escala estandarizada para la valoración de la intensidad de la inflamación de las uveítis posteriores (retinitis, coroiditis).
Exploraciones complementariasValoración estructuralLa tomografía de coherencia óptica (Optical Coherence Tomography [OCT]) (fig. 1E) es una prueba no invasiva que ofrece imágenes tomográficas de una resolución cuasi histológica de la mácula, permitiendo diagnosticar complicaciones con grave repercusión sobre la visión, como el edema macular (EM), los desprendimientos exudativos de retina y la neovascularización coroidea (NVC); permite asimismo cuantificar el grosor de la retina y por tanto monitorizar la respuesta terapéutica. Dado que requiere poco tiempo (2-3 minutos) y es muy sencilla de realizar se ha convertido probablemente en la exploración complementaria más importante en uveítis, especialmente si existe EM.
La angiografía fluoresceínica (AGF) permite estudiar la circulación retiniana, mientras que la angiografía con verde de indocianina (AVI) se emplea para el estudio de la circulación coroidea. La AGF puede mostrar alteraciones con hiperfluorescencia (fuga vascular indicativa de vasculitis de retina, tinción de lesiones inflamatorias, puntos de fuga en el epitelio pigmentario de la retina [EPR]) o con hipofluorescencia (cierre capilar e isquemia, bloqueo por lesiones inflamatorias); la AGF también permite diagnosticar el EM y la NVC.
La autofluorescencia del fondo ocular (AF) es una técnica de reciente introducción que capta la fluorescencia espontánea de las estructuras del fondo ocular que contienen lipofuscina (producto de degradación de los segmentos externos de los fotorreceptores, generada de forma fisiológica en las células del EPR). Dependiendo del proceso patológico, la cantidad de lipofuscina aumenta o disminuye, lo que condiciona respectivamente una señal de hiper o hipoautofluorescencia. La AF es especialmente útil en el diagnóstico y seguimiento de las uveítis posteriores que dañan la retina externa y el EPR, tales como la coroidopatía serpinginosa o la coroiditis multifocal (fig. 1F).
Valoración funcionalEl campo visual valora el daño del nervio óptico por glaucoma (complicación frecuente en pacientes con uveítis) y en los síndromes de manchas blancas (que pueden debutar con un escotoma temporal objetivable mediante esta prueba). La función del nervio óptico también la valoraremos mediante los potenciales evocados visuales. El electrorretinograma (ERG) mide la actividad eléctrica retiniana y puede estar alterado en algunos procesos en los que los hallazgos funduscópicos son sutiles o imperceptibles, como la coroidopatía en perdigonada, la retinopatía aguda zonal oculta externa (Acute Zonal Occult Outer Retinopathy [AZOOR]) y la retinopatía asociada al cáncer.
Historia clínica y exploración físicaEs fundamental elaborar una historia clínica completa y realizar una exploración física detallada, que junto con los hallazgos de la exploración oftalmológica permitan orientar de forma razonable el diagnóstico del paciente con uveítis8. Esta tarea será básicamente labor del internista. Desde un punto de vista práctico, debemos formularnos nuevamente 4 preguntas:
¿Cuáles son las características epidemiológicas del paciente?La edad es un dato particularmente importante, ya que en grupos de edad concretos (p. ej. en edad pediátrica) ciertas causas de uveítis son más frecuentes (tabla 2); el sexo y el origen étnico pueden ser también orientativos para el diagnóstico. Deberán recogerse siempre de forma detallada los antecedentes familiares, el consumo de fármacos, los antecedentes de traumatismo o cirugía ocular previa y la epidemiología relacionada con transmisión de enfermedades infecciosas8.
Uveítis: asociaciones epidemiológicas y clínicas
| Características demográficas y epidemiológicas | |
| Factor | Procesos con mayor riesgo |
| Edad menor de 15 años | |
| Edad: 0-5 años | Artritis idiopática juvenil, toxocariasis, neurorretinitis postviral, leucemia |
| Edad: 5-15 años | Artritis idiopática juvenil, pars planitis, toxocariasis, neurorretinitis postviral, sarcoidosis, leucemia |
| Origen étnico | |
| Nativo americano | Enfermedad de Vogt-Koyanagi-Harada |
| Japonés o coreano | Enfermedad de Vogt-Koyanagi-Harada, enfermedad de Behçet |
| Turco o mediterráneo | Enfermedad de Behçet |
| Procedencia | |
| Centro y Sudamérica | Cisticercosis |
| África subsahariana | Oncocercosis |
| Ingesta de carne cruda | Toxoplasmosis |
| Contacto con perros | Toxocariasis |
| Contacto con gatos | Toxocariasis, enfermedad por arañazo de gato, toxoplasmosis |
| Consumo de drogas por vía parenteral | Infección por VIH, endoftalmitis fúngica |
| Prácticas sexuales de riesgo | Infección por VIH, sífilis |
| Picaduras de garrapata | Enfermedad de Lyme |
| Hallazgos en la historia clínica y exploración física | |
| Signo/síntoma | Procesos asociados |
| Vitíligo, poliosis | Enfermedad de Vogt-Koyanagi-Harada |
| Eritema nudoso | Enfermedad de Behçet, sarcoidosis, tuberculosis |
| Pseudofoliculitis | Enfermedad de Behçet |
| Tromboflebitis superficial | Enfermedad de Behçet |
| Otras lesiones cutáneas específicas | Infección viral, sífilis, sarcoidosis, psoriasis, herpes zóster, herpes simple, enfermedad de Lyme, vasculitis, tuberculosis, lepra |
| Úlceras orales | Enfermedad de Behçet, enfermedad inflamatoria intestinal, celiaquía |
| Úlceras genitales | Enfermedad de Behçet, enfermedades de transmisión sexual |
| Aumento de tamaño salival o lagrimal | Sarcoidosis, linfoma |
| Xerostomía, xeroftalmia | Sarcoidosis, síndrome de Sjögren |
| Adenomegalias, esplenomegalia | Sarcoidosis, linfoma, tuberculosis, infección VIH |
| Diarrea | Enfermedad inflamatoria intestinal, enfermedad de Whipple |
| Tos, disnea, síntomas respiratorios | Sarcoidosis, tuberculosis, neoplasia |
| Sinusitis, rinorrea sanguinolenta | Granulomatosis de Wegener |
| Condritis nasal y auricular | Policondritis recidivante |
| Artritis | Artritis idiopática juvenil, síndrome de Reiter, artropatía psoriásica, sarcoidosis, artritis reumatoide, enfermedad de Behçet, enfermedad de Lyme, LES y otras conectivopatías, enfermedad inflamatoria intestinal |
| Sacroiliitis, lumbalgia, entesitis | Espondilitis anquilosante, artropatía psoriásica, síndrome de Reiter, enfermedad inflamatoria intestinal, espondiloartropatía indiferenciada |
| Cefalea, meningismo | Enfermedad de Vogt-Koyanagi-Harada |
| Manifestaciones neurológicas | Enfermedad de Vogt-Koyanagi-Harada, enfermedad de Behçet, sarcoidosis, esclerosis múltiple, linfoma del SNC |
| Sordera neurosensorial, vértigo | Enfermedad de Vogt-Koyanagi-Harada |
| Parálisis facial | Sarcoidosis |
| Quimioterapia, inmunosupresión | Infección por CMV, Candida y otros gérmenes oportunistas |
CMV: citomegalovirus; LES: lupus eritematoso sistémico; SNC: sistema nervioso central; VIH: virus de la inmunodeficiencia humana.
El objetivo del interrogatorio dirigido por aparatos es identificar síntomas sugerentes de enfermedad extraocular8. Muchas veces pueden existir manifestaciones clínicas que el paciente considera de poca relevancia o son poco sintomáticas, por lo que es recomendable realizar una anamnesis detallada incluso en pacientes que tras un interrogatorio rápido parecen estar asintomáticos. La lista de síntomas extraoculares que pueden sugerir una enfermedad sistémica es muy larga; los más relevantes se muestran en la tabla 2.
¿Existen hallazgos sugerentes de enfermedad extraocular en la exploración física?La exploración física completa puede mostrar signos sugerentes de enfermedad extraocular. Prácticamente cualquier hallazgo del examen físico puede ser relevante; probablemente sean los signos de afección mucocutánea, neurológica y del aparato locomotor los más orientativos en el paciente con uveítis (tabla 2)8.
¿Qué tratamiento ha recibido el paciente y cuál ha sido la respuesta?La respuesta al tratamiento recibido por el paciente aporta información sobre la intensidad de la uveítis y su causa. Es importante determinar si ha precisado o no corticoides, si los corticoides se han administrado a nivel ocular (administración tópica, subtenoniana o intravítrea) o por vía sistémica (y a qué dosis), si la respuesta ha sido completa, parcial o nula, o si se ha mantenido tras la reducción o suspensión del tratamiento. Ciertos patrones generales de respuesta son muy orientativos. Por ejemplo, una historia larga de episodios intermitentes con buena respuesta al tratamiento hace menos probable una etiología infecciosa o tumoral, mientras que es frecuente que las uveítis infecciosas mejoren inicialmente con el tratamiento antiinflamatorio aunque más tarde empeoren nuevamente; en el caso de los síndromes de enmascaramiento neoplásicos, la respuesta al tratamiento con corticoides suele ser escasa o nula, a excepción del linfoma vitreorretiniano que puede responder parcialmente.
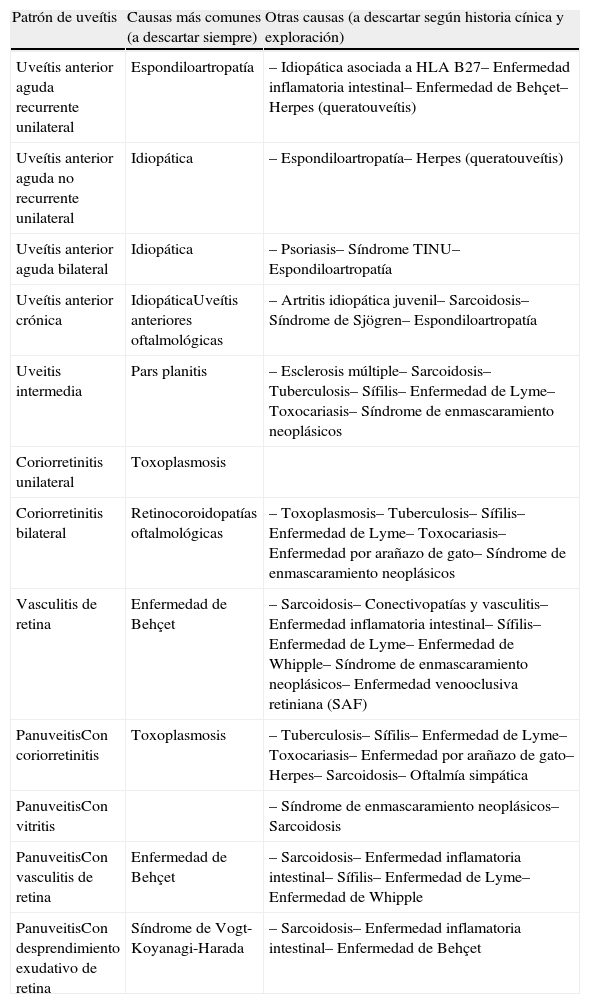
Diagnóstico diferencialNo existe una fórmula sencilla que permita responder a la típica pregunta «¿Qué le pido a una uveítis?». En función de los hallazgos de la exploración oftalmológica, del curso clínico y de la lateralidad de la uveítis pueden reconocerse 12 patrones clínicos (tabla 3). Cada patrón es causado con diferente frecuencia por diferentes procesos, por lo que considerando cuáles son más probables en función de la edad, epidemiología y signos y síntomas del paciente podremos plantear un plan diagnóstico razonable.
Aproximación diagnóstica según patrón de uveítis: Etiologías más frecuentes
| Patrón de uveítis | Causas más comunes (a descartar siempre) | Otras causas (a descartar según historia cínica y exploración) |
| Uveítis anterior aguda recurrente unilateral | Espondiloartropatía | – Idiopática asociada a HLA B27– Enfermedad inflamatoria intestinal– Enfermedad de Behçet– Herpes (queratouveítis) |
| Uveítis anterior aguda no recurrente unilateral | Idiopática | – Espondiloartropatía– Herpes (queratouveítis) |
| Uveítis anterior aguda bilateral | Idiopática | – Psoriasis– Síndrome TINU– Espondiloartropatía |
| Uveítis anterior crónica | IdiopáticaUveítis anteriores oftalmológicas | – Artritis idiopática juvenil– Sarcoidosis– Síndrome de Sjögren– Espondiloartropatía |
| Uveitis intermedia | Pars planitis | – Esclerosis múltiple– Sarcoidosis– Tuberculosis– Sífilis– Enfermedad de Lyme– Toxocariasis– Síndrome de enmascaramiento neoplásicos |
| Coriorretinitis unilateral | Toxoplasmosis | |
| Coriorretinitis bilateral | Retinocoroidopatías oftalmológicas | – Toxoplasmosis– Tuberculosis– Sífilis– Enfermedad de Lyme– Toxocariasis– Enfermedad por arañazo de gato– Síndrome de enmascaramiento neoplásicos |
| Vasculitis de retina | Enfermedad de Behçet | – Sarcoidosis– Conectivopatías y vasculitis– Enfermedad inflamatoria intestinal– Sífilis– Enfermedad de Lyme– Enfermedad de Whipple– Síndrome de enmascaramiento neoplásicos– Enfermedad venooclusiva retiniana (SAF) |
| PanuveitisCon coriorretinitis | Toxoplasmosis | – Tuberculosis– Sífilis– Enfermedad de Lyme– Toxocariasis– Enfermedad por arañazo de gato– Herpes– Sarcoidosis– Oftalmía simpática |
| PanuveitisCon vitritis | – Síndrome de enmascaramiento neoplásicos– Sarcoidosis | |
| PanuveitisCon vasculitis de retina | Enfermedad de Behçet | – Sarcoidosis– Enfermedad inflamatoria intestinal– Sífilis– Enfermedad de Lyme– Enfermedad de Whipple |
| PanuveitisCon desprendimiento exudativo de retina | Síndrome de Vogt-Koyanagi-Harada | – Sarcoidosis– Enfermedad inflamatoria intestinal– Enfermedad de Behçet |
TINU: síndrome de nefritis túbulo-intersticial y uveítis.
Fuente: modificado de Bañares A et al.6.
Son las más frecuentes, aproximadamente un 45-60% del total de las uveítis5–8.
Uveítis anterior aguda recurrente unilateral
Las espondiloartropatías son las causantes de casi la mitad de las uveítis anterior aguda recurrente unilateral (UAARU), característicamente con un cuadro de uveítis anterior de inicio agudo y con inflamación intensa; el segundo lugar en frecuencia lo ocupan las UAARU idiopáticas, asociadas a HLA B27 positivo o no5–8,11,12. La infección por herpes simple (y con menor frecuencia por virus varicela-zóster) puede causar una iridociclitis que curse con un patrón de UAARU, muchas veces derivada de la afectación corneal (queratouveítis), por lo que es común encontrar queratitis con infiltrados y cicatrices corneales; las uveítis herpéticas son habitualmente granulomatosas, suelen afectar siempre al mismo ojo, característicamente elevan la PIO y dan lugar a menudo a atrofia parcheada o sectorial del iris13–15. Es posible confirmar el diagnóstico mediante reacción en cadena de la polimerasa (PCR) en humor acuoso. En el caso del virus varicela-zóster, habitualmente (pero no siempre) suele existir el antecedente de herpes zóster en la rama oftálmica del trigémino. Otras causas menos frecuentes de las UAARU son la enfermedad inflamatoria intestinal (en especial colitis ulcerosa) y la enfermedad de Behçet, que al igual que las espondiloartropatías causa una uveítis anterior aguda con intensa inflamación16–19.
Uveítis anterior aguda no recurrente unilateralHabitualmente no son necesarias exploraciones complementarias en pacientes con un primer episodio de uveítis anterior aguda unilateral, ya que en la mayor parte de los casos se tratan de uveítis idiopáticas5–8. Únicamente realizaremos un estudio dirigido en aquellos casos en los que la anamnesis o la exploración física sean positivas, habitualmente orientado a descartar espondiloartropatía o queratouveítis herpética. Muchas veces este patrón es la forma de inicio del anterior, y el diagnóstico se realiza cuando se producen brotes sucesivos.
Uveítis anterior aguda bilateralEs un patrón infrecuente; la mayor parte de los casos son idiopáticos, aunque este patrón puede observarse en uveítis asociadas a psoriasis y raramente en los procesos descritos en los apartados anteriores y que habitualmente causan uveítis aguda unilateral5–8.
Aunque muy infrecuente, en los pacientes con uveítis anterior aguda bilateral hay que descartar el síndrome de nefritis túbulo intersticial y uveítis (Tubulo Interstitial Nephritis and Uveitis Syndrome, [TINU syndrome]), de etiología desconocida, que afecta habitualmente a adultos jóvenes. Las manifestaciones sistémicas (fiebre, artralgias, exantema, nefritis) suelen aparecer antes que la uveítis. Lo más característico es la presencia de nefritis túbulo-intersticial aguda, con deterioro variable de función renal, aumento urinario de beta-2-microglobulina, glucosuria normoglucémica y sedimento urinario patológico. La aparición de uveítis anterior aguda bilateral suele ser la clave para el diagnóstico; es importante el diagnóstico precoz, ya que la respuesta a corticoides habitualmente es buena20.
Uveítis anterior crónicaLa mayor parte se deben a procesos puramente oftalmológicos, como la ciclitis heterocrómica de Fuchs, el síndrome de Posner-Schlossman (crisis glaucomatociclíticas), las uveítis facogénicas, las uveítis infecciosas postquirúrgicas (frecuentemente por Propionibacterium acnes) y las asociadas a lentes intraoculares; o bien son uveítis idiopáticas (uveítis anteriores crónicas en las que no se identifica ningún cuadro puramente oftalmológico bien definido y con anamnesis y exploración física negativas para enfermedad extraocular)5–8. En estos pacientes no es necesaria la realización de exámenes complementarios. Recientemente se ha identificado un cuadro indistinguible del síndrome de Posner-Schlossman causado por citomegalovirus en pacientes inmunocompetentes, cuyo diagnóstico puede confirmarse mediante PCR en humor acuoso21.
Existen una serie de procesos sistémicos que son causantes de aproximadamente la quinta parte de los casos de uveítis anterior crónica; normalmente se encontrarán hallazgos característicos en la exploración ocular y/o manifestaciones clínicas extraoculares sugerentes y que suelen ser previas a la uveítis, por lo que puede orientarse el diagnóstico con la historia clínica y la exploración. Estos procesos son la artritis idiopática juvenil (la artritis precede casi siempre al desarrollo de la uveítis), el síndrome de Sjögren (xerostomía y xeroftalmía que asocia inflamación uveal) y la sarcoidosis (característicamente con precipitados retroqueráticos granulomatosos)5–8,21–26.
Uveítis intermediaAproximadamente el 4-8% del total de las uveítis son uveítis intermedias, aunque suponen aproximadamente el 20-25% de los casos remitidos a centros de referencia y de las uveítis diagnosticadas en niños y adolescentes5–8,27. La pars planitis es la causa más frecuente de uveítis intermedia (aproximadamente dos tercios de los casos); es una uveítis intermedia idiopática, con mínima inflamación en cámara anterior, iniciada en ocasiones en edad pediátrica y con mayor frecuencia en el adulto joven y caracterizada por vitritis con formación snowbanks o de snowballs, que con frecuencia se acompañan de flebitis en la retina periférica y edema macular quístico.
Además de la pars planitis, los dos procesos que con mayor frecuencia pueden causar uveítis intermedia son la sarcoidosis24–26 y la esclerosis múltiple (EM)28,29. A diferencia de la pars planitis, ocasionalmente la uveítis intermedia de la EM se asocia a una uveítis anterior, que puede ser granulomatosa; también se ha descrito una menor incidencia de EM en las uveítis intermedias asociadas a EM que en las pars planitis. Es motivo de discusión si es necesario realizar el despistaje de esclerosis múltiple en todos los casos de uveítis intermedia, ya que la uveítis intermedia puede preceder a las manifestaciones neurológicas; en nuestra opinión, solo debería realizarse el estudio en pacientes con manifestaciones neurológicas, en casos en los que la uveítis intermedia se asocia a uveítis anterior granulomatosa y no hay un diagnóstico etiológico alternativo (especialmente en mujeres de 20-45 años, teniendo en cuenta la epidemiología de la esclerosis múltiple) y en aquellos casos en los que se vaya a emplear terapia anti-TNF (contraindicada en la esclerosis múltiple).
Entre las causas menos frecuentes de uveítis intermedia se encuentran ciertas infecciones y parasitosis (tuberculosis, sífilis, enfermedad de Lyme, enfermedad de Whipple, toxocariasis) y los síndromes de enmascaramiento neoplásicos, que deben sospecharse cuando no hay respuesta al tratamiento antiinflamatorio adecuado, especialmente en personas mayores de 50 años (linfoma intraocular, melanoma coroideo, metástasis) y en niños (retinoblastoma)5–8. La tuberculosis y la sífilis son dos de las grandes simuladoras de uveítis, pudiendo causar prácticamente cualquier patrón de uveítis, por lo que es recomendable descartar estos dos procesos en el estudio de una uveítis intermedia30,31.
Uveítis posteriorSuponen aproximadamente el 10-15% de las uveítis en nuestro medio5–8.
Coriorretinitis unilateralCasi siempre este patrón es causado por la toxoplasmosis5–8, que habitualmente causa una coriorretinitis focal. El diagnóstico se realiza mediante la exploración en función de las características morfológicas de la lesión. Las formas multifocales o difusas son más raras, salvo en inmunodeprimidos. Es infrecuente la aparición de varios focos de coriorretinitis a la vez, pero la presencia de lesiones activas asociadas a cicatrices inactivas en el mismo ojo o en el contralateral es muy orientativa para el diagnóstico. La serología frente a Toxoplasma solo tiene utilidad como test confirmatorio. En un pequeño número de casos, las coriorretinitis unilaterales son debidas a otras infecciones y a formas unilaterales de coriorretinitis puramente oftalmológicas habitualmente bilaterales. Por último, merece la pena citar la necrosis retiniana aguda, una forma de coriorretinitis herpética (causada tanto por herpes simple como herpes zóster) que puede ser uni o bilateral (aunque habitualmente comienza de forma unilateral), y que se caracteriza por la presencia de uno o más focos de necrosis retiniana con vasculopatía oclusiva arteriolar, intensa vitritis y con frecuencia hemorragias y afectación del nervio óptico8,13.
Coriorretinitis bilateralEn más del 60% de los casos se corresponden con retinocoroidopatías exclusivamente oftalmológicas5–8: coroidopatía serpinginosa, coroidopatía en perdigonada (birdshot retinochoroidopathy), epiteliopatía pigmentaria placoide multifocal posterior aguda (EPPMPA), coroiditis multifocal con panuveítis, coroidopatía punteada interna, síndrome de los puntos blancos múltiples evanescentes, epitelitis pigmentaria retiniana aguda, retinopatía externa zonal aguda y síndrome de fibrosis subretiniana y uveítis. Algunas enfermedades sistémicas pueden simular algunos de estos síndromes oculares: la sífilis puede causar lesiones similares a la EPPMPA y la sarcoidosis similares a la coroidopatía en perdigonada. El diagnóstico depende del oftalmólogo, por lo que la labor del internista en la mayor parte de las ocasiones se centra en el control del tratamiento inmunosupresor que algunos de estos procesos precisan (especialmente la coroidopatía serpinginosa y la coroidopatía en perdigonada). En segundo lugar, tendremos que considerar las causas infecciosas: tuberculosis, sífilis, leptospirosis y enfermedad de Lyme (que suelen causar coriorretinitis multifocal), toxocariasis (más frecuente en niños, que puede causar coriorretinitis tanto focal como difusa), enfermedad por arañazo de gato (coriorretinitis focal o multifocal, y con frecuencia neurorretinitis con aparición de estrella macular), e infecciones oportunistas asociadas a infección VIH y otros estados de inmunodepresión8. En ocasiones, la tuberculosis puede simular un cuadro de coroidopatía serpinginosa (serpinginous-like), por lo que debe tenerse en cuenta en aquellos casos en los que la morfología de las lesiones sea atípica o no haya respuesta adecuada al tratamiento inmunosupresor32. Ocasionalmente, los síndromes de enmascaramiento neoplásicos pueden causar una coriorretinitis bilateral6–8.
Vasculitis de retinaAunque la vasculitis de retina puede encontrarse en una amplia variedad de procesos, la mayor parte (55-60%) son idiopáticas o asociadas a procesos puramente oftalmológicos (angeítis en rama escarchada, vasculitis de retina idiopática con aneurismas y neurorretinitis, vasculitis de retina multifocal aguda hemorrágica, pars planitis, retinocoroidopatía en perdigonada)8,33. La enfermedad de Eales es una forma de vasculitis retiniana muy característica, que afecta principalmente a varones jóvenes, y que se caracteriza por la presencia de vasculitis periférica con periflebitis que puede acompañarse de oclusión venosa, neovascularización retiniana periférica, hemorragias vítreas y retinianas recurrentes, y que en última instancia, puede causar un desprendimiento de retina por tracción; en un número significativo de pacientes con enfermedad de Eales se ha detectado mediante PCR la presencia de DNA de Mycobacterium tuberculosis en muestras de humor acuoso, vítreas y de membranas epirretinianas, lo que sugiere una asociación con la tuberculosis8.
La mayor parte de las enfermedades autoinmunes sistémicas pueden causar vasculitis de retina; habitualmente suelen existir manifestaciones extraoculares lo suficientemente evidentes como para que pueda orientarse el diagnóstico con la historia clínica y la exploración. En nuestro medio, la enfermedad de Behçet es responsable de un 20-25% de los casos; puede observarse también vasculitis de retina en sarcoidosis, lupus eritematoso sistémico (LES), síndrome de Sjögren, granulomatosis de Wegener y otras vasculitis asociadas a ANCA, poliarteritis nudosa, artritis reumatoide, polimiositis y dermatomiositis, arteritis de Takayasu, policondritis recidivante, enfermedad de Crohn y síndrome de Susac (microangiopatía cerebral, retiniana y coclear)6–8,17,18,24,26,30,31,33,34. Asimismo, la vasculitis de retina ha sido descrita en una amplia variedad de cuadros infecciosos (sífilis, tuberculosis, enfermedad por arañazo de gato, enfermedad de Lyme, toxoplasmosis, herpes simple y herpes zóster, enfermedad de Whipple, rickettsiosis, infección VIH) y neoplásicos (síndromes paraneoplásicos, linfoma ocular, leucemia aguda)8,30,31,33,34. Por último, hay que recordar que el síndrome antifosfolipídico, asociado o no a LES, puede simular una vasculitis al causar enfermedad venooclusiva en la retina35.
PanuveítisAproximadamente un 5-20% del total de uveítis pueden definirse como panuveítis5–8 (tabla 3). Las panuveítis son a menudo difíciles de clasificar, ya que pueden coexistir vitritis o diferentes formas de afectación del polo posterior (coriorretinitis, vasculitis retiniana, desprendimientos serosos de retina). Existen algunos procesos, como la sarcoidosis, la oftalmía simpática, algunas infecciones (tuberculosis, sífilis) y los síndromes de enmascaramiento neoplásicos, que deben tenerse en cuenta en el diagnóstico diferencial en todos los casos de panuveítis8,24,26,30,31,36,37. En pacientes seleccionados, en los que la respuesta al tratamiento sea inadecuada y exista riesgo de ceguera, puede ser necesario para el diagnóstico obtener muestras (mediante biopsia vítrea, vitrectomía o biopsia coriorretiniana) para estudio anatomopatológico y/o microbiológico.
La mayor parte de las panuveítis con coriorretinitis son debidas a infecciones (toxoplasmosis, tuberculosis, sífilis y otras espiroquetas, herpes, toxocariasis); entre las causas no infecciosas destacan las formas idiopáticas, la sarcoidosis y la oftalmía simpática. Cuando la panuveítis se asocia a vasculitis retiniana, las causas más habituales son similares a las de las uveítis posteriores con vasculitis de retina. Por último, en el caso de las panuveítis con desprendimiento exudativo de retina, el síndrome de Vogt-Koyanagi-Harada8,37 es la causa más frecuente, aunque también pueden observarse desprendimientos serosos de retina en algunos casos de sarcoidosis, oftalmía simpática, enfermedad de Behçet y uveítis asociada a enfermedad de Crohn.
La oftalmía simpática8,36 es una panuveítis poco frecuente, granulomatosa, no necrosante, bilateral y difusa, que se desarrolla tras un traumatismo quirúrgico o accidental en un ojo (denominado ojo excitador), y que tras un período de latencia habitualmente no superior a tres meses, causa la aparición de panuveítis bilateral. El ojo que no sufrió el traumatismo se denomina simpatizante. Habitualmente requiere de tratamiento inmunosupresor para el control de la inflamación ocular.
El síndrome de Vogt-Koyanagi-Harada8,37 es un proceso inflamatorio granulomatoso multisistémico, caracterizado por una respuesta autoinmune dirigida contra antígenos melanocíticos presentes en el ojo (retina e iris), piel y faneras, meninges y oído interno. Tiene un curso clínico característico en 4 fases: prodrómica (similar a una viriasis, con meningitis linfocitaria y manifestaciones auditivas), uveítica aguda (panuveítis bilateral con desprendimientos exudativos de retina y papilitis), crónica o de convalecencia (despigmentación y atrofia coriorretiniana) y crónica recurrente (uveítis anterior granulomatosa bilateral y complicaciones oculares. Al igual que la oftalmía simpática, requiere habitualmente de corticosteroides e inmunosupresores para su tratamiento.
Comentario del caso clínico y conclusionesLas manifestaciones clínicas que presenta la paciente (miodesopsias, disminución de agudeza visual) y los hallazgos de la exploración oftalmológica (vitritis bilateral, exudados en bancos de nieve, flebitis en retina periférica, edema macular quístico bilateral) son características de una uveítis intermedia. Aunque la causa más frecuente de uveítis intermedia es idiopática, este proceso cursa habitualmente sin afectación de cámara anterior (o afectación mínima) y sin manifestaciones extraoculares, circunstancias que no se dan en nuestro caso. Existen algunos datos clínicos que nos deben alertar sobre la posibilidad de que se trate de una uveítis intermedia secundaria a otro proceso: presencia de uveítis anterior granulomatosa, antecedente epidemiológico de contacto tuberculoso y sintomatología extraocular (tos persistente y artralgias). Entre las causas comunes de uveítis intermedia se encuentran la sarcoidosis (la uveítis anterior granulomatosa bilateral y la clínica respiratoria y articular podrían orientarnos hacia este diagnóstico), la esclerosis múltiple (el patrón de uveítis anterior granulomatosa y uveítis intermedia en una mujer de entre 20 y 45 años es muy sugerente; la ausencia de manifestaciones neurológicas no excluye el diagnóstico ya que la uveítis puede aparecer antes que la enfermedad desmielinizante) y la tuberculosis (existe el antecedente epidemiológico de contacto tuberculoso y síntomas respiratorios, por lo que debemos sospecharla). Sería necesario plantear un plan diagnóstico que permitiera confirmar o excluir estos tres procesos. En el caso que nos ocupa, la prueba de tuberculina resultó negativa y la tomografía computarizada torácica de alta resolución mostró la presencia de adenopatías hiliares y patrón micronodular pulmonar muy sugerentes de sarcoidosis; el diagnóstico definitivo de sarcoidosis se confirmó mediante biopsia transbronquial de una adenopatía subcarinal, que mostró granulomas no caseificantes, con tinción de Ziehl y cultivo para micobacterias negativos.
En definitiva, el diagnóstico de las uveítis precisa de una comunicación fluida entre el oftalmólogo y el internista. Cada especialidad aporta áreas de conocimiento diferentes, que hay que integrar, por lo que el desarrollo de consultas conjuntas es probablemente la forma más idónea de abordar el diagnóstico y tratamiento de las uveítis. Desde el punto de vista del especialista en Medicina Interna, es necesario familiarizarse con las técnicas utilizadas en la exploración oftalmológica y la terminología empleada en la descripción, diagnóstico y seguimiento evolutivo de las uveítis. La identificación del patrón clínico de la uveítis es la herramienta básica para la orientación diagnóstica; una vez identificado el patrón, el internista puede aportar al diagnóstico la visión integral del paciente, una historia clínica completa y una exploración física detallada, que permitirán decidir de forma razonable las pruebas complementarias necesarias.


