


ER-12. - SÍNDROME DE CHANARIN-DORFMAN EN 3 HERMANOS DE UNA FAMILIA SIN CONSANGUINIDAD
1Servicio de Medicina Interna. Hospital Comarcal de la Axarquía. Vélez-Málaga. 2Servicio de Dermatología, 3Servicio de Hematología, 4Servicio de Anatomía Patológica, 5Servicio de Alergología, 6Servicio de Aparato Digestivo. Hospital Regional Universitario de Málaga. Málaga.
Objetivos: El síndrome de Chanarin-Dorfman (SCD) es una rara enfermedad genética del metabolismo lipídico. El objetivo es describir la presentación clínica de este síndrome y su manejo diagnóstico-terapéutico.
Métodos: Descripción de 3 hermanos adultos que fueron diagnosticados de SCD, valorando sus características clínicas, las pruebas que ayudaron al diagnóstico y el tratamiento empleado.
Resultados: De los 3 pacientes, 2 eran varones (de 40 y 39 años) y una mujer (35 años), nacidos en una familia sin consanguinidad. Acudieron a la consulta externa de Dermatología por presentar una fina descamación cutánea sobre una base eritematosa que afectaba fundamentalmente a cara, abdomen, brazos y piernas y sequedad cutánea generalizada, desde los primeros años de vida, pero nunca lo habían consultado. En su historia clínica destacaban haber sido estudiado por hepatopatía desde hacía años, diagnosticándose de hepatoesteatosis en el caso del varón más mayor y la mujer. El segundo paciente varón refería historia de hepatopatía crónica en estadio cirrótico no filiado en seguimiento por digestivo. Además todos referían debilidad muscular generalizada de años de evolución. La mujer además presentaba desde hacía unos deterioro auditivo neurosensorial. A nivel analítico lo único que destacaba era un ligero aumento de las transaminasas con resto de resultados normales, incluyendo serología vírica y autoinmunidad. Desde consulta se les solicitó un frotis de sangre periférica con tinción de Giemsa poniéndose de manifiesto unos neutrófilos cargados de abundantes vacuolas lipídicas (Cuerpos de Jordan), características de esta enfermedad (fig.). En la biopsia muscular que se les realizó también se objetivaron depósitos lipídidos. Se solicitó el estudio genético para la detección de la mutación en el cromosoma 3 del Gen CG1 58 N209 confirmándose el diagnóstico. Los pacientes siguen tratamiento con emolientes cutáneos de urea para la ictiosis con aceptable resultado y el segundo paciente está en lista de espera de trasplante hepático y cumple tratamiento con ácido ursodeoxicólico, propanolol y vitamina E.
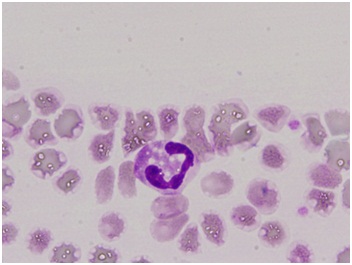
Discusión: El SCD es una rara enfermedad hereditaria autosómica recesiva, del metabolismo lipídico que se caracteriza por ictiosis y depósitos lipídicos a diferentes niveles (hepático, ocular, auditivo, músculo-esquelético y sistema nervioso central). Hasta el momento hay 58 casos descritos en la literatura, la mayoría del área mediterránea y oriente medio, y en población infantil. Los casos presentados son los de mayor edad diagnosticados hasta el momento.
Conclusiones: El SCD se trata de una enfermedad rara y sistémica, que debería ser tenida en cuenta por los clínicos, incluyéndose en el diagnóstico diferencial de la ictiosis. La presencia en un frotis de sangre periférica de neutrófilos con cuerpos de Jordan junto con clínica compatible puede ser suficiente para establecer su diagnóstico.


